入力ファイル
ネームリスト
CNTRL ネームリスト
CNTRL ネームリストは、計算全体の制御を行う入力。
キーワード |
デフォルト |
説明 |
|---|---|---|
Title |
'' |
計算のタイトル。 Viewer で出力必須項目。 |
ElecState |
'S1' |
計算する電子状態を指定。S1, S2, S3, D1, D2, D3, T1 が指定できるが現在は S1 のみ公開。 |
Method |
'MP2' |
計算方法を指定。HF, MP2, MP2D, MP3, LMP2, CAFI, CCPT が選択可能。XUFF が選択可能。Viewer で出力必須項目。 |
Gradient |
'NO' |
'YES' の場合、Gradient の計算を行う。主に MD に使用 (Geometory Optimization とは無関係)。 |
Memory |
1800 |
計算に使用するメモリの指定 (Mbyte)。 View で出力必須項目。 |
Natom |
0 |
原子の数を指定。ReadGeom を指定しない場合は必須。 |
Charge |
0 |
分子全体の電荷を指定。 |
ReadGeom |
'' |
分子の構造を記述したファイル。 通常は PDB 形式のファイルを指定する。これを指定しない場合は、座標データを直接入力する。 Viewer で出力必須項目。 |
WriteGeom |
'' |
計算結果を吐き出すファイル (CPF ファイル) を指定。 BioStation Viewer で使用。 拡張子に ajf を指定すると自動フラグメント分割機能によって分割されたフラグメント情報を含む ajf ファイルが出力される。この ajf ファイルを BioStation Viewer で読み込むことでフラグメントの分割を確認することができる。 Viewer で出力必須項目。 |
WriteIFIE |
'' |
機械学習などで必要となる最小限の計算結果が記載されたファイル (IFD ファイル) を吐き出す際に指定。 IFD ファイルの形式については、「補助ツール」 を参照のこと。 |
CpfVer |
10 |
cpf ファイルのバージョンを指定する。 デフォルトの 10 は Open1.0_rev10 に対応している。 Cpf ファイルフォーマットについては 「Check Point ファイル」 を参照のこと。Viewer で出力必須項目。 |
CPFBin |
'NO' |
'YES' の場合 CPF をバイナリファイルとして出力する。 |
Vector |
'OFF' |
Vector 型のプロセッサーの場合のみ使用 (NEC の SX など)。 |
THOVL |
1.00E-12 |
重なり積分値を用いた 2 電子積分のスクリーニング閾値。 |
E_THSWZ |
1.00E-12 |
シュワルツ不等式を用いた 2 電子積分のスクリーニング閾値。 指定した精度を保証する。 通常は THINTEG を与える。 |
G_THSWZ |
1.00E-12 |
シュワルツ不等式を用いた 2 電子積分 1 次微分のスクリーニング閾値。 指定した精度を保証する。 |
LogFile |
'' |
各 CPU における計算の進行状況を個別のファイルに書き出す。LOGFILE に指定したファイル名に「 _[Rank]」 を付けたファイルに書き出される。 NFS 以下を推奨。例: LogFile='/home/user/log/test'この場合、test_0, test_1 などのファイルが出来る。 |
Nprint |
3 |
全体の出力レベル。4 でダイマーのエネルギー、各 CPU ごとの時間などが出力される (非常に大きな出力ファイルとなるので注意されたい)。5 以上は、今は DEBUG 用なので指定しないほうが良い。0 にするとフラグメント分割のみ行って終了する。 |
TEIBUF |
-1 |
2 電子積分をバッファする領域を指定。 通常は指定しなくてよいが、メモリ容量が少ないサーバでの実行の場合、 0 に設定することを強く推奨する (ただし、毎回、積分計算を行うので 1 割程コストは上がる)。デフォルトでは Memory での指定値と同値が設定される。 |
FMOCNTRL ネームリスト
FMOCNTRL ネームリストは、FMO 計算の制御を行う入力。
キーワード |
デフォルト |
説明 |
|---|---|---|
FMO |
'ON' |
FMO 計算を行う。'OFF' の場合は conventional MO 計算となる。Viewer で出力必須項目。 |
(以下、FMO が 'ON' の場合のみ有効) |
||
Nbody |
2 |
FMO 計算における多体展開近似レベル。2/3/4: それぞれ 2 体、 3 体、 4 体を指定する。3 体、4 体を指定した場合には、後述の ESP 設定において ESPPTC='OFF' を指定すること。 |
FMO3 |
'ALL' |
Nbody=3 の場合に有効。'ALL', 'SL', 'L'が選択可能だが、sp2 分割を行う際には 'L' もしくは 'SL' を指定する。'SL' の方が高精度だが、その分計算コストも増加する。また、後述の ESP 設定において ESPPTC='OFF' を指定すること。sp2 分割の使用に際しては、節に示す点に留意されたい。 |
AutoFrag |
'ON' |
'ON' の場合、フラグメントの自動分割を行う。CNTRL ネームリストの ReadGeom で PDB ファイルを指定した場合のみ有効。'ON', 'OFF', 'HYBRID', 'READ' が指定可能。'HYBRID' の使用方法は「sp2 射影 BDA の使用方法」参照されたい。'READ'を指定する場合には、ReadFragList にてフラグメント情報を記載したファイル名を指定する。Viewer で出力必須項目。 |
ReadFragList |
'' |
AutoFrag で 'READ' を指定した際に、フラグメント情報が記載されたファイル名を指定する ('FragList.txt' など)。当該ファイルへのフラグメント情報の記載方法は &FRAGMENT ネームリストと同様である (ただし 1 行目の &FRAGMENT は不要)。 |
FragSizeResidue |
1 |
フラグメントの自動分割での分割残基数。AutoFrag で 'YES' を指定したときのみ有効。 |
FragSizeAminoacid |
'+amino' |
FMO 計算で使用。 自動フラグメント分割でアミノ酸を分割する場合に、側鎖でフラグメントに分割するかどうかを指定する。
Viewer で出力必須項目。 |
FragSizeNucleotide |
'/base' |
FMO 計算で使用。 自動フラグメント分割でヌクレオチドを分割する場合に、塩基をフラグメントに分割するかどうかを指定する。
|
Rsolv |
'' |
イオンが他のフラグメントにマージされるかどうかの判定距離を指定する。 例: Rsolv='Ca=2.8,Zn=2.4'現在指定できるイオンは Na, Mg, K, Ca, Mn, Fe, Co, Ni, Cu, Zn, Br の 11 種類となる。 Viewer で出力必須項目。 |
LigandCharge |
'' |
フラグメントごとにチャージを指定する。 通常の残基は、 AutoFrag が'ON' の場合、自動的に割り振られる。Viewer で出力必須項目。 |
NF |
0 |
フラグメントの数。AutoFrag が 'OFF' の場合は必須。 |
HybridFrag |
'' |
ハイブリッド分割の対象となるフラグメント番号を指定。 ここでのフラグメント番号は、最初に行う自動分割により付与れる番号。 AutoFrag='HYBRID' の場合は必須。例: HybridFrag='1,2,8-12' |
HybridNF |
0 |
ハイブリッド分割において、&FRAGMENT に記載されるフラグメントの数。HybridFrag で指定されたフラグメントが HybridNF 個のフラグメントに再分割される。AutoFrag='HYBRID' の場合は必須。 |
HybridSort |
'NO' |
ハイブリッド分割において、分割後にフラグメントの通し番号をソートするかを指定。 ソートは、各フラグメントに含まれる水素以外の原子の中で最も pdb 通し番号が小さな原子に着目し、昇順にフラグメントの並べ替えを行う。 |
MainProj |
'SP3' |
BDA に割り当てる射影演算子について、主として用いるものを指定する。
'SP2' がデフォルトとなる。&FRAGMENT の BDA BAA 設定箇所において、3 カラム目に 3 or 2 を指定することで、割り当てる射影演算子を上書き設定可能。詳細は「sp2 射影 BDA の使用方法」を参照。 |
LMOTyp |
'ANO' |
結合の切断に使用する Projection Orbital の種類を指定。'ANO', 'MOME', 'POPU' から選択。 |
ESPSET |
'Auto' |
環境静電ポテンシャルの設定を行う。'Manual' とすることで、以下の Laoc, Lptc に設定された値を用いた計算を実施する。 |
ESPAOC |
'ON' |
'ON' の場合、ESP-AOC 近似を有効にする。近似領域の始まりを Laoc で指定。 |
ESPPTC |
'ON' |
'ON' の場合、ESP-PTC 近似を有効にする。近似領域の始まりを Lptc で指定。 |
Laoc |
0 |
環境静電ポテンシャルを計算するときの近似レベル。 ファンデルワールス半径の倍数で表す。 フラグメント間の最近接の原子間の距離が、これよりも遠い場合は、3 中心積分で近似する (デフォルトでは、4 中心積分を使って計算しない)。* Viewer で出力必須項目。 |
Lptc |
2 |
環境静電ポテンシャルを計算するときの近似レベル。 ファンデルワールス半径の倍数で表す。 フラグメント間の最近接の原子間の距離がこれよりも遠い場合は、Mulliken Charge で近似する。* Viewer で出力必須項目。 |
esp_ptc_multipole |
'NO' |
'YES' の場合 esp-ptc 近似計算の代わりに CMM で計算する。 |
Max_order_multipole_ptc |
10 |
esp-ptc 近似計算を CMM で行う場合の、多重極モーメントの最大次数。 |
Ldimer |
2 |
ダイマー ES 近似を行う距離の閾値。 ファンデルワールス半径の倍数で指定。* Viewer で出力必須項目。 |
Dimer_es_multipole |
'NO' |
'YES' の場合、ダイマー ES 近似計算を CMM で行う。Viewer で出力必須項目。 |
Max_order_multipole |
10 |
ダイマー ES 近似計算を CMM で行う場合の、多重極モーメントの最大次数。 |
Ldimer_cmm |
5 |
ダイマー ES 近似計算を CMM で行う距離の閾値。 フラグメント間の最近接原子間距離がファンデルワールス半径の Ldimer_cmm 倍よりも離れているダイマーについて、CMM を適用する。Ldimer 倍より離れていて Ldimer_cmm 倍より近いダイマーは通常のダイマー ES 近似になる。Viewer で出力必須項目。 |
NP |
1 |
各フラグメントに割り振られる CPU コアの数。 各ノードの、1 CPU あたりのコア数を指定することを推奨する。 NP は、全体のコア数を割り切れる数でなければならない。Viewer で出力必須項目。 |
MaxSCCcyc |
250 |
モノマー SCC のサイクルの最大数。 |
MaxSCCenergy |
5.00E-07 |
SCC 収束判定のエネルギーの閾値 (Hartree)。 |
Write_SCC |
'' |
ファイル名を指定した場合、モノマー SCC の結果をファイルに出力する。 絶対パスで指定することを推奨。 |
Read_SCC |
'' |
ファイル名を指定した場合、モノマー SCC の結果をファイルから読む。 絶対パスで指定することを推奨。 |
Write_monomer_MP2 |
'' |
ファイル名を指定した場合、モノマー MP2 の結果をファイルに出力する。 絶対パスで指定することを推奨。 |
Read_monomer_MP2 |
'' |
ファイル名を指定した場合、モノマー MP2 の結果をファイルから読む。 絶対パスで指定することを推奨。 |
Write_dimer_es |
'' |
ファイル名を指定した場合、ダイマー ES の結果をファイルに出力する。 絶対パスで指定することを推奨。 |
Read_dimer_es |
'' |
ファイル名を指定した場合、ダイマー ES の結果をファイルから読む。 絶対パスで指定することを推奨。 |
Write_dimer |
'NO' |
'YES' の場合、ダイマー SCF およびダイマー MP2 の結果をファイルに出力する。Dimer_dir で指定されたディレクトリに出力する。ファイル名は、 dimer-result-[ifrag]-[jfrag]。 |
Read_dimer |
'NO' |
'YES' の場合、ダイマー SCF およびダイマー MP2 の結果をファイルから読む。Dimer_dir で指定されたディレクトリに Write_dimer が 'YES' の際に出力されたファイルが存在している必要がある。ファイルが存在しないもしくはファイルが読めないフラグメントペアに関しては、計算を行う。 |
Dimer_dir |
'' |
ダイマー計算の結果を出力するディレクトリを指定する。 このディレクトリは計算実行前にあらかじめ作成しておく必要がある (ファイルではないので注意する)。 |
SortDimer |
'YES' |
'YES' の場合、領域毎にダイマーリストをソートする。 |
SortDimerStackSize |
2048 |
ダイマーリストのソート (クイックソート) に使用できるスタックサイズをバイト数で指定する。 再帰呼び出し 1 回あたり 64B のスタックを使用します。ダイマー数がリストの最大長となるため、クイックソートの再帰呼び出しの最大数は となる。 現在、ダイマー数が 4 バイト整数であるため、使用される可能性のあるスタックサイズは 31*64=1984B。 ulimit コマンド等でシステムのスタックサイズを 2048B よりも十分大きく設定する必要がある。 |
WriteMOELevel |
'' |
出力するモノマーの軌道エネルギーレベルの指定。'ALL', 'HOMO:LUMO', 'HOMO-5:LUMO+5', 'HOMO', 'HOMO-5', 'LUMO', 'LUMO+5' など。デフォルトでは出力しない。 |
WriteMOEFrag |
'' |
軌道エネルギーを出力させるフラグメントの指定。 何も指定しない場合はすべてのフラグメントが対象となる。 例: '1-5,10-15' |
BalancedTaskLimit |
31250 |
並列計算の負荷均等分散のため計算タスクの計算量の概算値を降順にソートしたうえで計算ノードを決定する。 計算タスクの数が多い場合はこのソートが負担になる。 ソートを省略する閾値をここで指定する。 |
Pairs |
'' |
ダイマーリスト限定を行うフラグメントの指定。 |
PairsRegion |
'Center' |
ダイマー選択方法の指定。'All', 'Center', 'Group' から選択。BSSE ネームリストの Region 変数を参照。 |
PairsLnear |
0 |
近接するペアの有効距離 (ファンデルワールス半径の倍数)。 |
PairsLmax |
999 |
中心フラグメントからの有効距離 (ファンデルワールス半径の倍数)。 |
Note
* Laoc、Lptc、Ldimer などの閾値は各種 FMO 計算プログラムによって異なることがある。 GAMESS-FMO のなどの他の FMO 計算プログラムと計算結果と比較する際にはこれらの閾値を合わせる必要があることに注意する。
SCF ネームリスト
SCF を制御するためのネームリスト。コレスキー分解 (CDAM) 法の制御も SCF ネームリストで行う。
キーワード |
デフォルト |
説明 |
|---|---|---|
MaxSCFenergy |
1.00E-08 |
SCF 計算のエネルギーの収束の閾値。 |
MaxSCFdensity |
1.00E-06 |
SCF 計算の密度行列の収束の閾値。 |
MaxSCFcyc |
150 |
SCF 計算の最大反復回数。 |
DIISTYPE |
'C2_OLD' |
DIIS の手法を選択する。 以下が選択可能 ( C2_OLD が推奨であるが、SCF の収束が困難である場合には E+C1 など試みると良い)。
|
THINTEG |
1.00E-12 |
2 電子積分のスクリーニング閾値。 |
IFCD |
'NO' |
'YES' の場合、コレスキー分解 (CDAM) 法に基づく 2 電子積分近似を適用する。現在は MP2 法のみを加速。 GRADIENT='YES' の時は、MP2 微分計算で使用される 2 電子積分を CDAM によって近似する。Viewer で出力必須項目。 |
CDTYPE |
'Full' |
コレスキー基底の種類。
|
THCDINT |
1.00E-03 |
CDAM 近似におけるコレスキー基底選択のための閾値。 |
THCDLD |
1.00E-08 |
CDAM 近似における線形従属性判定のための閾値。 |
BASIS ネームリスト
基底関数を制御するためのネームリスト。
キーワード |
デフォルト |
説明 |
|---|---|---|
BasisSet |
'6-31G*' |
基底関数を指定する。 現在指定出来る基底関数は STO-3G, 3-21G, 6-31G, 6-31G*, 6-31G(d), 6-31G**, 6-31G(d,p), 6-31++G**, 6-31++G(d,p), 6-31G(C), 6-31G(d'), 6-31Gdag, 6-31G(d',p'), 6-31Gddag, 6-31G*(0.25), 6-31G**(0.25,0.15), 6-311G, 6-311G*, 6-311G**, 4-31G, cc-pVDZ。FMO 計算を行う場合は、結合を切断するための Projection Orbital が定義されている基底関数のみ使用可能。 なお、現在利用できる d 軌道はカーテシアン型 (6 成分) のみであり、球面調和型 (5 成分) は利用できない。 f 軌道 (対応するバイナリを使用) の場合もカーテシアン型 (10 成分) のみである。 'Read' を指定すると、ReadBS で指定された外部ファイルから基底関数を読み込む。Viewer で出力必須項目。 |
DiffuseOn |
'NO' |
'YES' の場合、特定の原子について、BasisSet で指定された 6-31G, 6-31G*, 6-31G** から 6-31G++G** に拡張。DiffuseOnElem, DiffuseOnFrag, DiffuseOnAtom を組み合わせた積集合によって適用原子を指定。 |
DiffuseOnElem |
'' |
DiffuseOn を適用する元素 (原子番号) の指定。O 原子と N 原子に適用する場合は DiffuseOnElem='7,8'。 |
DiffuseOnFrag |
'' |
DiffuseOn を適用するフラグメントの指定。 |
DiffuseOnAtom |
'' |
DiffuseOn を適用する原子 (通し番号) の指定。 |
OPTCNTRL ネームリスト
構造最適化計算に関するキーワードのネームリスト。
キーワード |
デフォルト |
説明 |
|---|---|---|
Opt |
'OFF' |
構造最適化計算の実行。 FMO 計算のみを実行する場合は 'OFF' を指定する。FMO 計算による各原子座標でのポテンシャルエネルギー勾配に基づき安定な原子構造を探索する場合は 'ON' を指定する。Viewer で出力必須項目。 |
OptFrag |
'' |
構造最適化するフラグメントを限定する場合の対象のリスト。 すべてのフラグメントの構造を最適化する場合は指定しない。 構造変化が重要な一部のフラグメントのみを構造最適化の対象とする部分構造最適化計算の場合は、それらのフラグメントの番号を列挙して指定する。 例: OptFrag='1,2,8-12' |
OptAtom |
'' |
構造最適化フラグメントから最適化する原子を限定するリスト。 対象のフラグメントに属するすべての原子の座標を最適化する場合は指定しない。 構造変化が重要な一部の原子のみを構造最適化の対象とする場合は、それらの原子の通し番号を指定する。 例: OptAtom='2-5,10-14, 18,20, 25,30,40,50' |
OptBDA |
'type1' |
部分構造最適化計算での最適化領域辺縁 BDA の最適化の選択。 最適化領域辺縁 BDA とは構造最適化の対象フラグメントの原子と対象でないフラグメントの原子との結合での片側(BDA 側)の原子のこと。 ペプチド結合ではアミノ基の Cα原子のこと。
OptBDA='type1' |
OptFrozenDomain |
'OFF' |
モノマー SCC 計算を凍結する FMO/FD 法の使用の指定。 FMO/FD 法を使用すると部分構造最適化の対象フラグメントから離れたフラグメントのモノマー SCC の更新が凍結され、計算時間が短縮される。
OptFrozenDomain='FD' |
Lbuffer |
2 |
FMO/FD 法での離れたフラグメントの判定距離。 FMO/FD 法を使用すると部分構造最適化の対象フラグメントから離れたフラグメントのモノマー SCC 計算が凍結される。 この離れたフラグメントを判定する距離を a.u. 単位 (Bohr 単位) で指定する。 例: Lbuffer = 3.0 |
OptDefrozenInterval |
10000 |
FMO/FD 法で凍結されたモノマー SCC 計算の解凍間隔。 FMO/FD 法を使用すると部分構造最適化の対象フラグメントから離れたフラグメントのモノマー SCC 計算は最適化の初回ステップで実施され、以降のステップでは凍結され実施されない。 しかし、指定したステップ間隔で凍結が解凍されモノマー SCC 計算が実施され、構造変化に追従するように更新される。 例: OptDefrozenInterval=10 |
OptFrozenDomainRestart |
'OFF' |
FMO/FD 法での継続再開時に凍結の保持を必須とする指示。 構造最適化の継続再開時には凍結フラグメントのモノマー SCC は一時的に解凍されて再計算されるが、 FMOCNTRL ネームリストの Read_SCC が指定されればモノマー SCC は再計算されずに凍結が保持され計算が継続される。
OptDefrozenInterval で指定の間隔のステップである場合は上記の指定に関わらず解凍される。使用例 |
OptMonomerEnergy |
'ALL' |
部分構造最適化計算での全エネルギーの計算法の選択。 全エネルギーにすべてのフラグメントのモノマー項を含める場合は 'all' を指定する。全エネルギーに構造最適化の対象でないフラグメントのモノマー項を含めずに構造最適化の対象のフラグメントのみでエネルギーの変化を確認する場合は 'partial' を指定する。例: OptMonomerEnergy='partial' |
EnergyBeforeOpt |
'OFF' |
最適化前にすべてのフラグメントによるエネルギー計算の実行。
EnergyBeforeOpt='ON' |
EnergyAfterOpt |
'OFF' |
最適化後にすべてのフラグメントによるエネルギー計算の実行。
EnergyAfterOpt='ON' |
OptMethod |
'BFGS' |
構造最適化に用いる原子座標の更新アルゴリズム。
'FRCG' (Fletcher-Reeves CG 法)、'PRCG' (Polak Ribere CG 法)、'HSCG' (Hestenes-Stiefel CG 法)、'DYCG' (Dai-Yuan CG 法)を選択できる。PRCG 法は FR CG 法より収束までの反復回数が少ない。 'CG' は 'FRCG' と同義。'BFGS': 過去のエネルギー勾配の履歴から近似計算したエネルギーの二階微分行列(ヘシアン)からエネルギー最小地点を予測して原子の座標を更新する方法 (Broyden-Fletcher-Goldfarb-Shanno 法)。メモリ使用量が多く自由度の大きい系への適応には注意。'Redundant': 原子間の結合長、結合角、二面角が系の座標の自由度の数より多く選ばれ、これら冗長な変数の更新結果が原子の座標に反映される方法。OptGDIIS との併用は不可。メモリ使用量が多く自由度の大きい系への適応には注意。例: OptMethod='Redundant' |
OptRedundantIntraFrag |
1.25 |
冗長内部座標としての原子間結合の判定に用いられる係数。OptMethod='Redundant' の指定による冗長内部座標を用いた構造最適化計算では、基準値より短い距離の原子間に結合長の内部座標が設定される。同一のフラグメントに属する原子間あるいは互いに結合するフラグメントに属する原子間では、両原子の元素半径の和に、この値を乗じた値が基準値となる。 例: OptRedundantIntraFrag=1.50 |
OptRedundantInterFrag |
2 |
冗長内部座標としての原子間結合の判定に用いられる係数。OptMethod='Redundant' の指定による冗長内部座標を用いた構造最適化計算では、基準値より短い距離の原子間に結合長の内部座標が設定される。互いに結合しないフラグメントに属する原子間では、両原子の元素半径の和にこの値を乗じた値が基準値となる。 例: OptRedundantInterFrag=2.50 |
OptGDIIS |
'NO' |
構造最適化計算の座標の更新での GDIIS 法の利用。 GDIIS 法は構造最適化計算の最適なステップ幅を計算する処理を GDIIS 外挿計算に置き換える。 ただし、Farkas-Schlegel 条件 (Phys. Chem. Chem. Phys., 2002, 4, 11-15) により、GDIIS を適用すべきか判定し、適合する場合は GDIIS 外挿を、不適合の場合は OptMethod で指定した SD/CG/BFGS 法(Redundant 法は指定不可)により更新座標を決定する。 しかし、顕著な効果が確認されているのは OptMethod='SD' の場合だけである。 |
GDIISTYPE |
'C1' |
DIIS 線形方程式解法の選択。 選択によって、DIIS 外挿計算に使用するベクトルが異なる。
'E' と 'E+C1' は、効果がほとんど確認されていない。'C1' と 'C2' の間に大きな差はないが、やや 'C1' の方が収束が早いように見受けられる。そのため、既定値 'C1' を変更する必要はほとんどないだろう。 |
NGDIIS |
4 |
GDIIS の履歴長。 DIIS 法は反復毎に一定のパラメータを保持し続けなければならない。 保持し続ける反復履歴の長さを NGDIIS で指定する。 値が増加するとメモリ使用量が増加する。 10 以上では、収束が遅くなる/収束しなくなる場合が多く、NGDIIS を大きくしたからといって収束性が良くなるわけではない。 分子構造に依存するが、3〜5 が適当であろう。 |
LPRINT_GDIIS |
0 |
GDIIS 用出力制御。 GDIIS に関してより詳細な出力を必要とする場合に設定する。 |
OptMaxStep |
0.3 |
最適化方向への原子座標の移動幅の最大許容値。OptMethod に 'SD'/'CG'/'BFGS' を指定した場合は各原子が最適化方向に少しずつ移動されながらエネルギーの変化が調べられる。この原子の移動の幅の最大許容値を a.u.単位 (Bohr 単位) で指定する。 例: OptMaxStep=0.1 |
OptLineSearch |
'cubic' |
最適化方向でのエネルギーの最小点の予測法。OptMethod に 'SD'/'CG'/'BFGS' を指定した場合の最適化方向でのエネルギーの変化は移動幅の多項式で補間され、その最小点に原子座標が更新される。その多項式の種類を指定する。
OptLineSearch='quadratic' |
OptConvergence |
'Default' |
構造最適化の反復計算の収束判定の各基準値の一括指定。 基準値には MaxGrad, RmsGrad, MaxDisp, RmsDisp があり、すべての基準が満たされて収束と判定される。各基準の値は個別にも指定できるが、各推奨値をこの値で一括して指定できる。 指定法と設定される各基準値は以下の表を参照。 'Default' を選択あるいは何も指定しない場合には各基準は個別に指定された値もしくはそれらのデフォルト値となる。 指定例 |
MaxGrad |
1.00E-02 |
構造最適化の収束判定の基準 (hartree/bohr)。 エネルギー勾配の最大値に対する基準値を指定する。 |
RmsGrad |
7.00E-03 |
構造最適化の収束判定の基準 (hartree/bohr)。 エネルギー勾配の二乗平均平方根に対する基準値を指定する。 |
MaxDisp |
2.00E-02 |
構造最適化の収束判定の基準 (bohr)。 原子座標の移動幅の最大値に対する基準値を指定する。 |
RmsDisp |
1.40E-02 |
構造最適化の収束判定の基準 (bohr)。 原子座標の移動幅の二乗平均平方根に対する基準値を指定する。 |
MaxCyc |
100 |
構造最適化の反復回数の上限値。 構造最適化計算の反復計算が収束しない場合の反復を中止する反復の上限回数を指定する。 |
OptOutput |
'' |
構造最適化途中での分子構造を保存するファイル名。 指定可能なファイル形式は以下のとおり。
|
OptHistory |
'' |
構造最適化計算の履歴を記録するファイル名。 拡張子は .goh。 このファイルに各原子のエネルギー勾配、座標、BFGS 法の場合のヘッセ行列が記録される。 継続計算時にこのファイルが読み込まれると厳密な継続計算となる。 |
OptRestart |
'OFF' |
構造最適化計算の前回の出力ファイルからの継続再開の指示。 新規に構造最適化計算を実行する場合は 'OFF' を指定する。この場合は ReadGeom のファイルの座標を初期構造として構造最適化計算が開始される。OptOutput、OptHistory、FMOCNTRL ネームリストの Write_SCC で指定されるファイルに継続計算のための各種データが記録される。前回の構造最適化計算で出力された各種ファイルから構造最適化計算をさらに進める場合は 'ON' を指定する。OptInput、OptHistory、FMOCNTRL ネームリストの Read_SCC で指定されるファイルから各種データが入力され計算が継続される。 例: OptRestart='ON' |
OptInitialSCC |
'ON' |
構造最適化計算でのモノマー SCC の収束値の再利用の指定。 構造最適化の各ステップで前ステップの構造でのモノマー SCC の収束値を今ステップの構造でのモノマー SCC の初期値とすることで収束までの計算時間を短縮する場合は 'ON' を指定する。各ステップでのモノマー SCC を最初から計算する場合は 'OFF' を指定する。例: OptInitialSCC='ON' |
OptInput |
'' |
構造最適化計算の再開のための分子構造が記録されたファイル名。 指定可能なファイル形式は以下のとおり。
|
Example
-
1 2 3 4 5 6 7 8
&FMOCNTRL Read_SCC='monomer.scc' Write_SCC='monomer.scc' / &OPTCNTRL OptRestart='ON' OptFrozenDomainRestart='ON' /
Note
-
MaxGradRmsGradMaxDispRmsDisp'VeryLoose'1.0e-2 0.7e-2 1.0e0 1.0e0 'Loose'1.0e-2 0.7e-2 2.0e-2 1.4e-2 'Tight'1.0e-3 0.7e-3 2.0e-3 1.4e-3 'VeryTight'0.45e-3 0.30e-3 1.80e-3 1.20e-3 'Default'- - - -
Example
-
1 2 3 4 5
OptConvergence='Default' MaxGrad=1.0e-2 RmsGrad=0.7e-2 MaxDisp=2.0e-2 RmsDisp=1.4e-2
SCZV ネームリスト
2 体 FMO-HF レベルの Gradient 計算に追加補正する応答項を制御するためのネームリスト。
キーワード |
デフォルト |
説明 |
|---|---|---|
DimerResponseTerm |
'NO' |
ダイマー応答項の有効化。 |
Debug |
'NO' |
デバッグモードで実行。 |
SCZVMaxCycle |
100 |
SCZV の最大反復回数。 |
SCZVThreshold |
1.00E-08 |
SCZV の終了判定閾値。 |
MP2 ネームリスト
MP2 計算を制御するためのネームリスト。
キーワード |
デフォルト |
説明 |
|---|---|---|
NP_MP2_IJ |
1 |
IJ バッチの並列化の分割の数。 ij 駆動型エンジン用。 でなければならない。 |
NP_MP2_S |
0 |
S バッチの並列化の分割の数。 でなければならない。 0 を指定すると になる。 |
MemoryMP2 |
0 |
MP2 計算で使用するメモリサイズ (Mbyte)。0 を指定すると、Memory から計算される空きメモリサイズがデフォルトとなる。 |
IFSCS |
'YES' |
'YES'だとスピン成分スケーリング (SCS) による修正 MP2 エネルギーも計算する。 |
OSSCAL |
1 |
ユーザー定義の SCS-MP2 の反平行スピンのスケーリングファクター。IFSCS='READ' の場合のみ有効。 |
PSSCAL |
1 |
ユーザー定義の SCS-MP2 の平行スピンのスケーリングファクター。IFSCS='READ' の場合のみ有効。 |
NBODY |
2 |
FMOCNTRL ネームリストの NBODY の値を 、MP2 ネームリストの NBODY の値を とすると、HF 法を N 体項まで、MP2 法を M 体項まで考慮する FMO 計算を行う。ただし あり、 で FMO(N)-MP2 法、 では FMO(N')-MP2 法となる。 デフォルトは 。 現在は FMO3 のみ有効で、FMO4 については対応中。 |
CHKFZC |
'YES' |
'YES' の場合には深い占有軌道を省く (Frozen Core)。 |
CHKFZV |
'YES' |
大きな軌道エネルギーを持つ仮想軌道を省く (Frozen Virtual) |
THRFZV |
1.d4 |
仮想軌道のカットオフ (CGKFZV) のエネルギー閾値 |
FZCINP |
'' |
凍結軌道数を元素ごとに指定 例: 'Na=1,Ca=5' |
LPRINT |
2 (3) |
MP2 関連の出力レベル。デフォルト値を推奨。 |
IFPRNM |
'YES' |
'YES' の場合には、PRMP2 計算を行う。 |
MP2DNS ネームリスト
キーワード |
デフォルト |
説明 |
|---|---|---|
NP_MP2D_I |
1 |
I バッチに関する並列度。 デフォルト値を推奨。 |
NP_MP2D_S |
0 |
S バッチに関する並列度。 デフォルト値を推奨。 |
IFCPHF |
'NO' |
CPHF を使用し、より高精度な MP2D 計算を行う。 |
MP2GRD ネームリスト
キーワード |
デフォルト |
説明 |
|---|---|---|
ITCPHF |
'YES' |
CPHF 方程式を解くかどうか。 |
MAXITR |
60 |
CPHF 方程式を解く場合の最大繰り返し数。 |
MemoryMP2Grad |
0 |
0 を指定すると、Memory から計算される空きメモリサイズがデフォルトとなる |
MP3 ネームリスト
MP3 計算を制御するためのネームリスト。 MP2.5 計算を行う場合は Method='MP3' とした上で下記のスピンのスケーリングファクター SCL2OS、SCL2PS、SCL3 に 1、1、0.5 を指定する。
キーワード |
デフォルト |
説明 |
|---|---|---|
NP_MP3_IJ |
1 |
IJ バッチに関する並列度。 |
NP_MP3_S |
0 |
S バッチに関する並列度。 デフォルト値を推奨。 0 を指定すると NP/NP_MP3_IJ になる。 |
MemoryMP3 |
0 |
MP3 計算で使用するメモリサイズ (Mbyte)。0 を指定すると、Memory から計算される空きメモリサイズがデフォルトとなる。 |
IFSCS |
'YES' |
Grimme の SCS-MP3 エネルギーを計算するフラグ。 |
IFMANU |
'NO' |
手入力で SCS スケーリングパラメータを入れるフラグ。 |
SCL2OS |
1 |
SCS-MP2 の反平行スピンのスケーリングファクター。 デフォルトは MP2.5。 Grimme の SCS-MP3 の場合は 6/5=1.2 を指定。 |
SCL2PS |
1 |
SCS-MP2 の平行スピンのスケーリングファクター。 デフォルトは MP2.5。 Grimme の SCS-MP3 の場合は 1/3=0.333... を指定。 |
SCL3 |
0.5 |
SCS-MP3 の平行スピンのスケーリングファクター。 デフォルトは MP2.5。 Grimme の SCS-MP3 の場合は 1/4=0.25 を指定。 |
CHKFZC |
'YES' |
'YES'の場合には深い占有軌道を省く (Frozen Core)。 |
CHKFZV |
'YES' |
大きな軌道エネルギーを持つ仮想軌道を省く (Frozen Virtual)。 |
THRFZV |
1.d4 |
仮想軌道のカットオフ (CGKFZV) のエネルギー閾値。 |
FZCINP |
'' |
凍結軌道数を元素ごとに指定。 例: 'Na=1,Ca=5' |
LPRINT |
2 |
MP3 関連の出力レベル。 |
LMP2 ネームリスト
Local MP2 計算 (FILM 解析) を制御するためのネームリスト。
キーワード |
デフォルト |
説明 |
|---|---|---|
IJBATCH |
1 |
IJ バッチ数。 |
LOC |
'POPU' |
局在化方法の指定。'POPU' または 'MOME'。 |
CHKFZC |
'YES' |
'YES' の場合、Frozen Core を適用する。 |
FZCINP |
'' |
凍結軌道数を元素ごとに指定。 例: 'Na=1,Ca=5' |
BSGRID |
'' |
BioStation 用のデータを書き出すファイル名を指定する。 絶対パスで指定することを推奨。 LMP2 計算が行われた全てのダイマーペアに関して、別々のファイルに出力される。 このファイルが無ければ、軌道レベルの解析ができない。 |
LRD ネームリスト
局所応答分散相互作用の評価機能を制御するためのネームリスト。なお、本機能の一部には SMASH (http://smash-qc.sourceforge.net/) の数値積分ルーチンを使用している。
キーワード |
デフォルト |
説明 |
|---|---|---|
Order |
10 |
LRD の次数を指定。6, 8, 10 から選択可能。 |
Precision |
'Medium' |
LRD 計算に用いるグリッド等の細かさを指定。'Low', 'Medium', 'High' が選択可能。 |
Excl_BDA |
'NO' |
LRD 計算において、BDA の寄与を含めるかどうかを指定する。 |
ANALYSIS ネームリスト
解析用の設定を行う。
キーワード |
デフォルト |
説明 |
|---|---|---|
PIEDA |
'YES' |
PIEDA を実施するかどうかを指定する。 |
PrintNeighbor |
'NO' |
BDA/BAA で隔てられた隣のフラグメントとの相互作用エネルギーを出力するかどうかを指定する。 |
BSSE ネームリスト
Counterpoise 法による BSSE 補正計算を制御するためのネームリスト。
キーワード |
デフォルト |
説明 |
|---|---|---|
CP |
'OFF' |
'ON' の場合、IFIE に対する BSSE-CP 補正値の計算を行う。 |
Region |
'ALL' |
計算対象とするフラグメントペアの指定。 実際計算されるものは dimer-ES 近似の対象にならないペアのみ。
|
Lnear |
999 |
近接するペアの有効距離 (ファンデルワールス半径の倍数)。Region='Center' または Region='Group' を指定した場合に、有効距離内のすべての近接フラグメントペアも計算対象にする。 |
Lmax |
999 |
中心フラグメントからの有効距離 (ファンデルワールス半径の倍数)。Region='Center' の場合、Fragment とともに指定する。 |
Fragment |
'' |
計算対象となるフラグメントの指定。Region='Center' または Region='Group' の場合は必須。複数指定可能。 |
Method |
'' |
計算方法の指定 ('HF', 'MP2', 'MP3' など)。指定しない場合は本計算から継承される。 ただし MP2 の density 指定は解除される。 |
NPair |
0 |
読み込ませるフラグメントペアの数。Region='Read' の場合に必須。 |
ReadPair |
'' |
フラグメントペアを記述した外部ファイルの指定。FRAGPAIR ネームリストで指定された形式で記述。Region='Read' 指定が必須。 |
EnvESP |
'ON' |
'ON'の場合、環境静電場の影響を考慮する。 |
LPrint |
3 |
出力レベル。 |
Ghost |
'' |
Basis set superposition error (BSSE)の counterpoise 補正を行う場合の ghost atom または ghost fragment を指定する (80 文字以内)。 指定した数字は、HF 計算の場合は原子の番号と、FMO 計算の場合はフラグメントの番号と解釈される。 複数の原子/フラグメントを指定する場合は、',' (半角カンマ) で区切る。 連続した原子/フラグメントを指定する場合は、'-' (半角ハイフン) を使用する。 例: Ghost='1,2,10-20' |
Note
Counterpoise 法を用いた IFIE () に対する BSSE 補正の結果は、ログファイルには IFIE セクションに続けて出力される。出力形式は IFIE セクションと同様で、その値は Hartree-Fock エネルギーおよび電子相関エネルギーに対応した BSSE 値 ()である。最終的に補正された IFIE () を得るには、次の式で算出する必要がある。
Example
IFIE テーブル内において、dimer-es 近似の有無をそれぞれの対象となるフラグメントペアに対して T (True) および F (False) と表記するのに加え、後者でさらに BSSE 補正対象となるフラグメントペアに対して BSSE テーブル内では B (BSSE) と表記する。 BSSE 補正計算の出力サンプル (水 5 量体を用いた BSSE 補正 (FMO2-MP2/6-31G**)) を以下に示す。
-
主な指定変数:
&CNTRL:Method='MP2'&FMOCNTRL:FMO='ON'&BASIS:BasisSet='6-31G**'&BSSE:CP='ON'Region='ALL'
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
## BSSE for non-bonding MP2-IFIE Frag. Non-bonding interaction energy / hartree / kcal/mol / kJ/mol 1 0.0084025237 5.272663 22.060823 2 0.0082862136 5.199677 21.755451 3 0.0085642502 5.374148 22.485436 4 0.0085948483 5.393349 22.565771 5 0.0086348014 5.418420 22.670668 ## BSSE for MP2-IFIE IJ-PAIR DIST DIMER-ES HF-IFIE MP2-IFIE PR-TYPE1 GRIMME JUNG HILL / A APPROX. / Hartree / Hartree / Hartree / Hartree / Hartree / Hartree ---------------------------------------------------------------------------------------------------- 2 1 1.746397 B 0.001813 0.001985 0.000000 0.001912 0.001876 0.000954 3 1 3.644075 B 0.000123 0.000124 0.000000 0.000110 0.000103 0.000078 3 2 1.734458 B 0.001917 0.002098 0.000000 0.002025 0.001988 0.001001 4 1 3.644147 B 0.000123 0.000132 0.000000 0.000118 0.000110 0.000083 4 2 3.672773 B 0.000117 0.000127 0.000000 0.000113 0.000107 0.000080 4 3 1.733246 B 0.001929 0.002118 0.000000 0.002043 0.002006 0.001011 5 1 1.736474 B 0.001957 0.002146 0.000000 0.002071 0.002033 0.001024 5 2 3.698206 B 0.000114 0.000115 0.000000 0.000103 0.000097 0.000072 5 3 3.656132 B 0.000123 0.000133 0.000000 0.000118 0.000111 0.000083
FRAGPAIR ネームリスト
BSSE 補正計算を行うフラグメントペアの指定を行うためのネームリスト。ペアとなるフラグメント番号を 5 桁右詰 (2I5) で入力し、ペアごとに改行を行う。
1 2 3 4 5 6 | |
SOLVATION ネームリスト
連続溶媒モデル (PB) を制御するためのネームリスト。
キーワード |
デフォルト |
説明 |
|---|---|---|
EFFECT |
'OFF' |
'ON' の場合、溶媒効果を課す。HF または MP2 に対応。 |
ITRMOD |
'Normal' |
反復方法の指定。 現在は閾値による収束判定 ( 'Normal') のみ有効。 |
MAXITR |
20 |
Grand iteration の反復回数の上限値。 |
THICNV |
1E-05 |
収束判定のための閾値。 |
SaveStat |
'' |
Grand iteration の履歴を保存するファイルを指定。 ファイル形式は Solvation Archived File (SAF) で拡張子は .saf。 収束前であれば途中状態からの、収束後であれば収束状態でのリスタート計算が可能。 また収束条件を変化させることで、新たな反復計算を行なうことも可能。 |
LoadStat |
'' |
SAF ファイルの指定。 リスタート計算を行なう。 |
IGSCC |
'Core' |
Grand iteration において Monomer SCC 計算の初期軌道として前ステップの結果を読み込む設定。
|
IniEHF |
'OFF' |
'ON' の場合、気相計算において高次相関計算を行わない。 |
IniCPF |
'' |
気相計算の結果を書き出す CPF のファイル名を指定。 |
PRBRAD |
1.4 |
溶媒の Probe 半径の指定 (Å 単位)。デフォルトは水溶媒。 |
EPSOUT |
80 |
溶媒の誘電率の指定。 デフォルトは水溶媒。 |
EPSIN |
1 |
溶質の誘電率の指定。 |
SRFTNS |
0.0072 |
溶媒和エネルギーの非静電項を表面積 (SASA) から計算するための表面張力係数 (kcal/mol / Å3 単位)。 |
SRFOFF |
0 |
溶媒和エネルギーの非静電項を表面積 (SASA) から計算するための補正値 (kcal/mol 単位)。 |
NSPHER |
1000 |
表面積 (SASA) の生成時の原子あたりの点の数。 |
Example
以下、Chignolin に対する FMO-PB 計算 (FMO2-MP2/6-31G レベル) を例に、溶媒計算における具体的な出力と値の見方について述べる。ジョブが流れ始めると、本計算に先立ち PB 計算の設定概要が出力される。このセクションにおいて、使用される原子半径を確認することができる。PBEQ ネームリストの RADSCL を用いたスケーリングを行う場合は特に注意する。
-
設定概要
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33
################################################################################ ## ## ## COMPUTATIONAL SUMMARY FOR INCLUDING SOLVENT EFFECT ## ## ## ################################################################################ ## OVERVIEW * RESTART [DISABLE] * STEP BEFORE GRAND ITERATION - Gas phase calculation (as 0th step) = YES - Electron density (atomic charges on solute) = HF - Correlation calculation = YES - Write check point file (gas phase) = NO * STEP IN GRAND ITERATION - Iteration in monomer-SCC loop for 1st step = NO - Reload previous results as guess for SCC = ON MEMORY - Electron density (atomic charges on solute) = HF - Total energy (judgement of convergence) = HF - The last step with correlation calculation = NO * STEP AFTER GRAND ITERATION - Correlation calculation = YES * RESULT - Write solvation free energy = YES - Write check point file (solvation) = YES - Save state of solvation for restart = YES ## ATOMIC RADIUS (DELPHI MODIFIED) / Angstrom H 1.150 C 1.900 N 1.600 O 1.600Note
## ATOMIC RADIUS (DELPHI MODIFIED) / Angstrom以下に、使用される原子半径が表示される。
本計算に移行すると、溶質と溶媒間の自己無撞着性を得るための大反復計算が開始される。ただし、第 0 ステップは gas phase の FMO 計算となる。各大反復ステップは以下に示すようなタイトルと収束状況を示す出力で区切られる。
-
途中経過
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
################################################################################ ## ## ## GRAND ITERATION FOR SOLVATION WITH HF DENSITY ## ## ( NUMBER OF STEP = 1 OF MAXIMUM NUMBER = 20 ) ## ## ## ################################################################################ **************************************************************** START FRAGMENT MOLECULAR ORBITAL CALCULATION **************************************************************** ##SOLV: Iteration = 1 < 20 ##SOLV: Total energy = -3798.3578162838 ##SOLV: Difference = 0.5310205832 > 0.0000100000 ##SOLV: Not converged yet. The iteration is continued.Note
- 上記は、大反復の 1 ステップを示している。
NUMBER OF STEP = 1は反復回数を示している (第 0 ステップは、gas phase 計算)。- 収束判定は、
##SOLV: Differenceに表示される。
- 収束は
THICNV変数によって制御されるため、大反復回数を減じるにはこの変数の値を十分に大きく設定する必要がある (MAXITR変数はあくまで大反復回数の上限値であり、上限値に達した時点でTHICNVを満たしていない場合、未収束となることに注意)。 - 収束に到達すると、大反復計算の収束履歴ならびに溶媒和自由エネルギーの出力が行われる。
- 上記は、大反復の 1 ステップを示している。
-
収束結果および溶媒和エネルギー
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38
################################################################################ ## ## ## SUMMARY OF SOLVATION CALCULATION ## ## ## ################################################################################ ## SUMMARY OF ITERATIONS FOR ELECTROSTATIC SOLVATION ENERGY Iter. Total Energy / Hartree Difference / Hartree ------------------------------------------------------ 1 -3798.3578162838 -0.5310205832 2 -3798.4095948533 -0.0517785695 3 -3798.4182447984 -0.0086499452 4 -3798.4200203659 -0.0017755674 5 -3798.4204431649 -0.0004227990 6 -3798.4205551472 -0.0001119824 7 -3798.4205871810 -0.0000320337 8 -3798.4205968919 -0.0000097110 ------------------------------------------------------ * Solvation calculation was successfully converged. ## SOLUTE TOTAL ENERGY SCF / Hartree MP2 corr. / Hartree Total / Hartree FMO2 in vacuo -3797.82679570 -7.65562513 -3805.48242083 in solvent -3798.42059689 -7.59127535 -3806.01187224 difference -0.59380119 0.06434979 -0.52945141 ( ES correction -0.67179481 ) ## SOLVATION FREE ENERGY FMO2 / Hartree FMO2 / kcal/mol Electrostatic -0.59380119 -372.61587016 Nonpolar 0.01231924 7.73044061 Total -0.58148195 -364.88542955Note
## SOLUTE TOTAL ENERGY:FMO2 in vacuoのTotal / Hartreeは を示している。differenceのTotal / Hartreeは を示している。
## SOLVATION FREE ENERGY:Nonpolarは を示している。
-
タンパク質受容体–リガンド複合体間の結合自由エネルギーを算定する場合には、複合体 (com)、受容体 (rec)、リガンド (lig) に対して同様な計算を行い、次のような関係式による算定が可能である。
- es 成分は FMO-PB カップリングから、np 成分は溶媒露出表面積 (SA) から経験的に見積もられる。
- 収束に到達した際、最終ステップのフラグメント相互作用解析 (IFIE または PIEDA) を確認する。この例題で注目するアミノ酸残基は荷電フラグメントである Gly(1), Asp(3), Glu(5) である。 Gly(1) は荷電した N 末端 (NH3+) を持つ。
-
IFIE と遮蔽効果
1 2 3 4 5 6 7 8 9 10 11 12
## PIEDA IJ-PAIR ES EX CT+mix Solv(ES) DI(MP2) q(I=>J) / kcal/mol / kcal/mol / kcal/mol / kcal/mol / kcal/mol / e ----------------------------------------------------------------------------------------------------- 3 1 -32.069720 -0.006986 -0.582390 28.022028 -0.625165 -0.000146 4 1 4.746559 0.000000 0.000000 -4.917634 0.000000 0.000000 4 2 1.104088 0.755338 -1.295819 -0.712827 -2.581517 0.008573 5 1 -20.495766 0.000000 0.000000 22.230499 0.000000 0.000000 5 2 1.403397 0.022265 0.314631 1.064433 -0.229299 0.000205 5 3 42.080606 8.154225 -4.512533 -40.527549 -4.390232 0.040620 6 1 2.301244 0.000000 0.000000 2.909690 0.000000 0.000000- 荷電フラグメント同士の相互作用 (3-1, 5-1, 5-3) は大きな静電 (ES) 成分を持つ一方で、それを打ち消すように溶媒による遮蔽効果 (Solv (ES)) が得られることで、総和として抑制される。遮蔽効果は、フラグメント同士がある程度離れており、なおかつお互いに溶媒面に露出している場合に大きな効果が得られることに注意する。
- ログには溶媒の静電項 (Solv (ES)) のみ出力されるが、CPF には非静電項 (Solv (NP)) も出力されるので BioStation Viewer を通して溶媒効果の総和を評価可能である。非静電項もログに出力する場合は
SOLVATIONネームリストにLPrint=4を指定する。
PBEQ ネームリスト
Poisson-Boltzmann 方程式ソルバーを制御するためのネームリスト。
キーワード |
デフォルト |
説明 |
|---|---|---|
MAXITR |
1000 |
連立方程式解法における反復回数の上限値。 |
JDGCNV |
'RMS' |
収束判定条件。 静電ポテンシャルの変化の最大値で判定する場合は'Max' 、全体の RMS 値で判定する場合は'RMS' を選択。 |
THRCNV |
1.00E-05 |
収束判定閾値の指定。 |
ATMRAD |
'DELPHI' |
接触半径の指定。'DELPHI', 'UFF', 'VDW' から選択可能。
'VDW' では Gaussian04 で用いられた距離情報が用いられる。個別元素ごとにマニュアル指定も可能。 例: ATMRAD='H=1.0,C=1.5' |
RADSCL |
1 |
ATMRAD で指定された原子半径に対してスケーリングを行う。 収束が上手くいかない場合に調整すると良い。 |
POP ネームリスト
密度解析を制御するためのネームリスト。
キーワード |
デフォルト |
説明 |
|---|---|---|
NBOANL |
'ON' |
'ON'の場合、NBO 解析を行う。Natural Population Analysis (NPA)に基づく原子電荷を出力する。 |
NBOFRG |
'' |
より詳細な NBO 解析を行うフラグメントの指定。 例: NBOFRG='1,2,3-5' |
ESP_PTC |
'MK' |
PTC 近似に用いる電荷を指定する。'RESP' とすることで、RESP 電荷を PTC 近似に用いることができる。ただし、以下の ESPFIT, ESPTYP と設定を統一しなければならない。 |
ESPFIT |
'OFF' |
'ON' の場合、静電ポテンシャル (ESP) フィッティングを行い、ESP 電荷の値を出力する。 |
ESPTYP |
'MK' |
ESP 電荷の種類。
|
Note
- NBO 解析ならびに ESP フィッティングの結果は Mulliken 密度解析セクションに続けて出力される。
- NBO 解析では、conventional MO 計算時には原子電荷に加え、自然軌道ごとの占有数や自然電子配置などの詳細情報が出力される。
- FMO 計算時は多体展開ごとの原子電荷が出力されるが、特に注目するフラグメントについては
NBOFRGで指定することによって、conventional MO 計算時と同等の詳細情報が得られる。
Example
-
- 主な指定変数
&CNTRL:METHOD='HF'&FMOCNTRL:FMO='OFF'&BASIS:BasisSet=6-31G'&POP:NBOANL='ON'
Conventional MO 計算による glycine に対する NBO 解析 (HF/6-31G)
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42
========================================================== ## NATURAL POPULATION ANALYSIS -- Ver.2.70(20120905) ========================================================== ## NATURAL ATOMIC ORBITAL POPULATION ------------------------------------------------- No. Atom NAO Type Occupancy ------------------------------------------------- 1 1 N 1s Core 1.999541 2 1 N 2s Valence 1.383815 3 1 N 3s Rydberg 0.000990 4 1 N 2px Valence 1.481290 5 1 N 3px Rydberg 0.002725 : (省略) ## SUMMARY OF NATURAL POPULATION ANALYSIS -------------------------------------------------------------------------------- Atom Res Frag Charge MCP Core Valence Rydberg Total -------------------------------------------------------------------------------- 1 N 1 1 -0.830545 0.0 1.999541 5.824288 0.006716 7.830545 2 H 1 1 0.461052 0.0 0.000000 0.537420 0.001529 0.538948 3 H 1 1 0.490658 0.0 0.000000 0.506040 0.003302 0.509342 4 H 1 1 0.490658 0.0 0.000000 0.506040 0.003302 0.509342 5 C 1 1 -0.350875 0.0 1.999186 4.335598 0.016090 6.350875 6 H 1 1 0.267899 0.0 0.000000 0.730716 0.001384 0.732101 7 H 1 1 0.267899 0.0 0.000000 0.730716 0.001384 0.732101 8 C 1 1 0.852060 0.0 1.999516 3.093676 0.054747 5.147940 9 O 1 1 -0.802408 0.0 1.999744 6.796504 0.006160 8.802408 10 O 1 1 -0.846399 0.0 1.999829 6.842590 0.003981 8.846399 -------------------------------------------------------------------------------- Total 0.000000 0.0 Core orbital 9.997816 ( 99.97816% of 10 ) Valence orbital 29.903587 ( 99.67862% of 30 ) Natural Minimal Basis (NMB) 39.901403 ( 99.75351% of 40 ) Natural Rydberg Basis (NRB) 0.098597 ( 0.24649% of 40 ) -------------------------------------------------------------------------------- Atom Res Frag Natural electron configuration -------------------------------------------------------------------------------- 1 N 1 1 [core]2s( 1.38)2p( 4.44)3p( 0.01) 2 H 1 1 1s( 0.54) : (省略) - 主な指定変数
-
FMO 計算による水 4 量体に対する NBO 解析 (FMO2-MP2/6-31G*)
- 主な指定変数
&CNTRL:METHOD='MP2'&FMOCNTRL:FMO='ON'&BASIS:BasisSet='6-31G*'&POP:NBOANL='ON'NBOFRG='2'
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56
========================================================================= ## NATURAL POPULATION ANALYSIS WITH MP2 DENSITY -- Ver.2.70(20120905) ========================================================================= ## SUMMARY OF NATURAL POPULATION ANALYSIS FOR SPECIFIED FRAGMENTS # NATURAL AO POPULATION -------------------------------------------------------- NAO Atom Res Frag AngMom Type Occupancy -------------------------------------------------------- 1 4 O 2 2 s Cor.( 1s) 1.999792 2 4 O 2 2 s Val.( 2s) 1.737655 3 4 O 2 2 s Ryd.( 3s) 0.005115 4 4 O 2 2 s Ryd.( 4s) 0.000564 5 4 O 2 2 px Val.( 2p) 1.685951 (省略) -------------------------------------------------------------------------------- Atom Res Frag Charge MCP Core Valence Rydberg Total -------------------------------------------------------------------------------- 4 O 2 2 -0.996747 0.0 1.999792 6.924778 0.072178 8.996747 5 H 2 2 0.481241 0.0 0.000000 0.516587 0.002172 0.518759 6 H 2 2 0.515587 0.0 0.000000 0.477888 0.006525 0.484413 -------------------------------------------------------------------------------- Total 0.000081 0.0 ( Formal charge: 0.0 ) Core orbital 1.999792 ( 99.98958% of 2 ) Valence orbital 7.919252 ( 98.99066% of 8 ) Natural Minimal Basis (NMB) 9.919044 ( 99.19044% of 10 ) Natural Rydberg Basis (NRB) 0.080875 ( 0.80875% of 10 ) -------------------------------------------------------------------------------- Atom Res Frag Natural electron configuration -------------------------------------------------------------------------------- 4 O 2 2 [core]2s( 1.74)2p( 5.19)3s( 0.01)3p( 0.04)3d( 0.03) 5 H 2 2 1s( 0.52) 6 H 2 2 1s( 0.48)2s( 0.01) -------------------------------------------------------------------------------- ## NATURAL ATOMIC POPULATIONS ---------------------------------------------------- Atom Res Frag Charge Charge Pop FMO2-HF FMO2-MP2 FMO2-MP2 ---------------------------------------------------- 1 O 1 1 -1.024136 -0.996755 8.996755 2 H 1 1 0.537066 0.515507 0.484493 3 H 1 1 0.486948 0.481110 0.518890 4 O 2 2 -1.024171 -0.996747 8.996747 5 H 2 2 0.487092 0.481241 0.518759 6 H 2 2 0.537137 0.515587 0.484413 7 O 3 3 -1.024172 -0.996784 8.996784 8 H 3 3 0.487111 0.481257 0.518743 9 H 3 3 0.537076 0.515528 0.484472 10 O 4 4 -1.023966 -0.996610 8.996610 11 H 4 4 0.487073 0.481231 0.518769 12 H 4 4 0.536942 0.515435 0.484565 - 主な指定変数
-
ESP フィッティング
- ESP フィッティングでは、conventional MO 計算時に原子電荷の値に加え、フィッティング時の統計情報やフィッティング電荷による多重極子などが出力される。
- FMO 計算時は、多体展開ごとの原子電荷のみを表示する。
-
- 主な指定変数
&CNTRL:METHOD='HF'&FMOCNTRL:FMO='OFF'&BASIS:BasisSet='6-31G*'&POP:ESPFIT='ON'ESPTYP='MK'
Conventional MO 計算によるメタノール CH3OH に対する ESP フィッティング (HF/6-31G*)
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53
============================================================ ## ELECTROSTATIC POTENTIAL FITTING -- ver.1.0 (20120329) ============================================================ ------------------ FITTING INFOMATION ------------------ ESP-FITTED CHARGE TYPE = Merz-Kollman SAMPLING SCHEME = Merz-Singh-Kollman scheme NUMBER OF SAMPLING POINTS = 429 FITTING METHOD = Normal fitting FIXED CHRAGE CENTERS = 0 UNIQUE UNFROZEN CENTERS = 6 ITERATIONS = 0 (<= 20) CONVERGENCE THRESHOLD = 0.000E+00 (<= 0.100E-04) Num Atom Charge Equiv Fixed Initial Rstrnt.Wt. ----------------------------------------------------------- 1 H 0.058110 0 F 0.000000 0.000000 2 C 0.195785 0 F 0.000000 0.000000 3 O -0.669999 0 F 0.000000 0.000000 4 H 0.427075 0 F 0.000000 0.000000 5 H -0.005485 0 F 0.000000 0.000000 6 H -0.005485 0 F 0.000000 0.000000 ----------------------------------------------------------- Total: 0.000000 ------------------ FITTING STATISTICS ------------------ INITIAL CHARGE SUM = 0.000000 (AVE. = 0.000000) INITIAL CHARGE SUM OF SQUARES = 0.000000 (AVE. = 0.000000) ESP-FITTED CHARGE SUM = -0.000000 (AVE. = -0.000000) ESP-FITTED CHARGE SUM OF SQUARES = 0.673060 (AVE. = 0.334928) ESP(QM ) SUM = -0.170835 (AVE. = -0.000398) ESP(QM ) SUM OF SQAURES [SSVPOT] = 0.116817 (AVE. = 0.016502) ESP(PTC) SUM = -0.235268 (AVE. = -0.000548) ESP(PTC) SUM OF SQUARES = 0.114994 (AVE. = 0.016372) RESIDUAL SUM ( ESP(PTC)-ESP(QM ) ) = -0.064433 (AVE. = -0.000150) RESIDUAL SUM OF SQUARES [CHIPOT] = 0.001823 STANDARD ERROR [sqrt(CHIPOT/N)] = 0.002061 RELATIVE RMS [sqrt(CHIPOT/SSVPOT)] = 0.124913 --------------------- DIPOLE MOMENT (Debye) --------------------- X = -1.51656 Y = 1.05424 Z = -0.00000 Tot = 1.84699 -------------------------------------------- TRACELESS QUADRUPOLE MOMENT (Debye*Angstrom) -------------------------------------------- QXX = 2.47244 QYY = 0.51530 QZZ = -2.98774 QXY = 4.02817 QYZ = -2.86432 QZX = -2.86432 - 主な指定変数
-
FMO 計算による水 4 量体に対する ESP フィッティング (FMO2-MP2/6-31G*)
- 主な指定変数
&CNTRL:METHOD='MP2D'&FMOCNTRL:FMO='ON'&BASIS:BasisSet='6-31G*'&POP:ESPFIT='ON'ESPTYP='MK'
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
============================================================================= ## ELECTROSTATIC POTENTIAL FITTING WITH MP2 DENSITY -- ver.1.1 (20120905) ============================================================================= ## ESP-FITTING TYPE: Merz-Kollman ----------------------------------------- Atom Res Frag Charge Charge FMO2-HF FMO2-MP2 ----------------------------------------- 1 O 1 1 -0.885462 -0.862145 2 H 1 1 0.449552 0.432844 3 H 1 1 0.437338 0.430642 4 O 2 2 -0.891628 -0.868306 5 H 2 2 0.439360 0.432638 6 H 2 2 0.452298 0.435677 7 O 3 3 -0.887149 -0.863682 8 H 3 3 0.439334 0.432547 9 H 3 3 0.446399 0.429635 10 O 4 4 -0.887349 -0.864040 11 H 4 4 0.438074 0.431303 12 H 4 4 0.449234 0.432886 - 主な指定変数
GRIDCNTRL ネームリスト
- 電子密度などのグリッドファイルを出力するためのネームリスト。
- 出力できるグリッドファイルの種類:
- 分子軌道 (.mo)
- 電子密度 (.den)
- CNS 形式の電子密度 (.cns)
- 静電ポテンシャル (.esp)
- 等電子密度面上の静電ポテンシャル (.map)
- 等電子密度面上の電場ベクトル (.efv)
- 自然軌道対 (.pno)
- .mo 及び .den はファイル I/O を介さず出力できるが、その他のグリッドファイルは FMO の計算結果を中間ファイルに保持する必要があるため、FMO 計算実行時に
FMOCNTRLネームリスト等において中間ファイルの出力先を指定する必要がある (詳細は実行例を参照)。
キーワード |
デフォルト |
説明 |
|---|---|---|
GRID |
'NO' |
'YES' の場合、電子密度などのグリッドデータが作成される。Viewer で出力必須項目。 |
WriteMO |
'NO' |
'YES' の場合、 WriteMORegion で指定されたフラグメントの分子軌道 (Molecular Orbital) のグリッドデータをファイルに出力する。MP2 計算をした場合も HF レベルの分子軌道が出力される。 |
WriteMORegion |
'' |
WriteMO に 'YES' が指定された場合、分子軌道のグリッドデータを出力するフラグメントをフラグメント番号で指定する。例: WriteMORegion='1,2,8-12' |
MOlevel |
'Homo:, Lumo' |
WriteMOGrid に 'YES' が指定された場合、出力される MO の範囲を指定する。指定には、以下に示す方法がある。
|
WriteDen |
'NO' |
'YES' の場合、電子密度 (Electron density) のグリッドデータをファイルに出力する。出力されるファイルの拡張子は .den となる。 電子密度の単位は 1/bohr3 で出力される。 Method='MP2D' の場合、MP2 レベルの電子密度が出力される。多体の FMO 計算 (FMO3 、FMO4) を行った場合、多体 FMO に基づく電子密度が出力される。 |
WriteEsp |
'NO' |
'YES' の場合、静電ポテンシャル (Electrostatic potential) のグリッドデータをファイルに出力する。出力されるファイルの拡張子は .esp となる。 |
WriteMap |
'NO' |
'YES' の場合、等電子密度面上の静電ポテンシャルのグリッドデータをファイルに出力する。出力されるファイルの拡張子は .map となる。 |
WriteEfv |
'NO' |
'YES' の場合、等電子密度面上の電場ベクトルのグリッドデータをファイルに出力する。出力されるファイルの拡張子は .efv となる。 |
WritePNO |
'NO' |
'YES' の場合、 CAFI によって得た自然軌道対 (Pair Natural Orbital) のグリッドデータをファイルに出力する。出力されるファイルの拡張子は .pno となる。 Method='CAFI' によって CAFI 計算を実行する場合にのみ指定できる。 |
IsosurfaceValue |
0.001 | WriteMapGrid または 'WriteEfvGrid' に 'YES' が指定された場合、静電ポテンシャルまたは電場ベクトルを計算する等電子密度面の値を指定する。 |
PartialGrid |
'NO' |
'YES' の場合、分子の部分構造に対するグリッドデータを計算する。'NO' のままだと分子全体に対するグリッドデータを計算する。 |
PartialGridRegion |
'' |
PartialGrid に 'YES' が指定された場合、グリッドデータの計算対象となる部分構造をフラグメント番号で指定する。例: PartialGridRegion ='1,2,8-12' |
BaseName |
'' |
出力されるグリッドデータファイルのベースネームを指定する。 指定されない場合は、読み込んだ CPF ファイルのベースネームを基に決定される。 |
AutoGridBoxSize |
'YES' |
'YES' の場合、入力分子が収まるようなグリッドボックスのサイズを GridDeltaX と GridBoxSpace の値を基に自動で設定する。PartialGrid='YES' の場合、PartialGridRegion で指定されたフラグメントが収まるようなグリッドボックスが設定される。 |
GridDeltaX |
0.25 |
グリッドデータのメッシュの細かさを Å 単位で指定する。 値を小さくするほど得られるグリッドデータがきめ細かくなるが、ファイルサイズが大きくなる。 |
GridBoxSpace |
3 |
グリッドデータを計算するボックスのサイズに関する指定。 ボックスは分子の大きさに合わせて自動的に計算され、 GridBoxSpace では分子の端とボックスの最短距離を Å 単位で指定する。通常はデフォルトのままで良いと考えられる。 |
nx ny nz |
0 |
グリッドボックスの x, y, z 軸方向のメッシュ数。AutoGridBoxSize が 'NO' の時に有効。 |
xmin xmax ymin ymax zmin zmax |
0 |
グリッドボックスの x, y, z 軸方向の開始点および終止点。AutoGridBoxSize が 'NO' の時に有効。 |
WriteDenCNS |
'NO' |
'YES' なら CNS 形式の電子密度グリッドを出力する。拡張子は.cns となる。 電子密度の単位は 1/Å3 で出力される。 Method='MP2D' の場合、MP2 レベルの電子密度が出力される。多体の FMO 計算(FMO3 、FMO4)を行った場合、多体 FMO に基づく電子密度が出力される。 |
AutoGridBoxSizeCNS |
'YES' |
'YES' なら分子全体が収まるような直方体のグリッドボックスを GridDeltaXCNS と GridBoxSpaceCNS の値を基に自動で設定する。PartialGrid='YES' の場合は、PartialGridRegion で指定されたフラグメントが収まるようなグリッドボックスが設定される。'NO' の場合、以下に示す CNS 形式のパラメーター (格子定数など) でグリッドボックスを指定することができる。このオプションは WriteDen_CNS にのみ有効となる。 |
GridDeltaXCNS |
0.25 |
CNS 形式のグリッドデータのメッシュの細かさを Å 単位で指定する。 値を小さくするほど得られるグリッドデータがきめ細かくなるが、ファイルサイズが大きくなる。 |
GridBoxSpaceCNS |
3 |
CNS 形式のグリッドデータを計算するボックスのサイズに関する指定。 ボックスは分子の大きさに合わせて自動的に計算され、 GridBoxSpaceCNS では分子の端とボックスの最短距離を Å 単位で指定する。 |
nanbnc |
0 |
CNS フォーマットにおける Na、Nb、Nc。 La/Na、Lb/Nb、Lc/Nc が a, b, c 軸方向の刻み幅 (、、) となる。 |
lalblc |
0 |
CNS フォーマットにおける La/Na、 Lb/Nb、 Lc/Nc (Å)。 La/Na、Lb/Nb、Lc/Nc が a, b, c 軸方向の刻み幅 (、、) となる。 |
aminamaxbminbmaxcmincmax |
0 |
CNS フォーマットにおける a, b, c 軸方向の開始点と終止点を整数で指定する。 (0,0,0)が原点に対応する。 |
alphabetagamma |
90 |
結晶格子定数の α、β、γ (degree) |
Example
-
-
FMO-HF レベルで.den グリッド及び.mo グリッドを出力する場合の入力例:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
&CNTRL Method='HF' ReadGeom='/path/to/file/sample.pdb' WriteGeom='/path/to/file/sample.cpf' / &FMOCNTRL FMO='ON' / &GRIDCNTRL GRID='YES' WriteDen='YES' WriteMO='YES' WriteMORegion='1' MOlevel='Homo-5:Lumo+5' /- .den 、.cns について
- FMO 計算時にダイマーリスタートファイルなどの中間ファイルの指定無しに出力できる。
- 多体 FMO (FMO3 、FMO4) 計算時は多体 FMO に基づく電子密度が出力される。
- MP2D 計算時は MP2D に基づく電子密度、MP2D (CPHF) 計算時は MP2D (CPHF) に基づく電子密度が出力される。
- .mo について
- FMO 計算時にダイマーリスタートファイルなどの中間ファイルの指定無しに出力できる。
- モノマーの分子軌道なので多体 FMO を指定しても結果は変わらない。
- MP2 計算時も HF レベルの分子軌道が出力される。
- .den 、.cns について
-
.esp 、.map 、.efv グリッドを出力する場合の入力例:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17
&CNTRL Method='HF' ReadGeom='/path/to/file/sample.pdb' WriteGeom='/path/to/file/sample.cpf' / &FMOCNTRL FMO='ON' Write_dimer='YES' Dimer_dir='/path/to/file/sample_dir/' Write_SCC=''/path/to/file/sample.scc' / &GRIDCNTRL GRID='YES' WriteMap='YES' WriteEsp='YES' WriteEfv='YES' /- . esp 、.map 、.efv について
- FMO 計算実行時に中間ファイル (cpf 、モノマー SCC 、ダイマーリスタートファイル)を指定する必要がある。
- FMO2-HF レベルでのみ計算できる(多体 FMO (FMO3 、FMO4) または MP2D と組み合わせて指定できない)。
- . esp 、.map 、.efv について
-
Note
- その他の補足事項
- SCF 計算が未収束なフラグメントが存在する場合はグリッドデータは出力されないので注意する。
- PartialGrid は中間ファイルを経由してグリッドデータを出力する場合のみ有効。
MCP ネームリスト
MCP (Model Core Potential) を利用した計算を行うためのネームリスト。
キーワード |
デフォルト |
説明 |
|---|---|---|
CHKMCP |
'NO' |
'YES' の場合、MCP を用いる。 |
NELEM |
0 |
MCP を用いる元素の種類を入力。 2 種類の元素に MCP を適用する場合には NELEM=2 とする。現時点では原子番号 3-20, 31-38, 49-55, 58, 78, 80-86 が内蔵されている。 |
ELEM |
'' |
MCP を適用する元素の原子番号を入力。炭素と窒素に適用する場合には ELEM='6,7' とする。 |
ReadMCP |
'' |
MCP 用のパラメータセットファイル。 内蔵されていない元素に関しては、以下のサイトから GAMESS-US 形式で MCP 用のパラメータセットを取得し、後述の修正を加えたファイル名をここで指定することで MCP を適用することが可能である。例: ReadMCP='MCP.dat' |
Note
外部からMCPパラメータを読み込むためのファイルは、GAMESS-US形式でダウンロードした後、以下のように元素名を追記する必要があるので注意されたい。
-
例:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
!****************************************************************************** ! Element : Ca ! Basis : MCP+NOSeC-V-DZP = MCP relativistic (661/52) ! Effective Charge : 10.0 ! Term : 1S Valence configuration : 1s(2)2p(6)2s(2) ! SCF energy : -36.38052312 a.u. ! Valence Correlation energy : -0.02473 a.u. ! Reference ! Authors : H. Anjima, S. Tsukamoto, H. Mori, H. Mine, M. Klobukowski, E. Miyoshi, [inner and valence shell] ! Journal : J. Comput. Chem. 28, 2424-2430 (2007) ! Authors : M. Sekiya, T. Noro, Y. Osanai, T. Koga, [valence correlated set] ! Journal : Theor. Chem. Acc. 106, 297 (2001) !****************************************************************************** Calcium MCP READ s 6 1 151.5755200 0.1479950 : (省略)- 14 行目に元素名を追加した (上記の例では
Calcium)。
- 14 行目に元素名を追加した (上記の例では
CAFI ネームリスト
- CAFI 解析を制御するためのネームリスト。
CNTRLネームリストにおいてMethod='CAFI'を指定した場合に有効。
キーワード |
デフォルト |
説明 |
|---|---|---|
FRAGCAFI |
'' |
解析を行うフラグメントの指定。 例: FRAGCAFI='1,2,3-5' |
METLOC |
'PIPE' |
局在化手法の指定。'PIPE','BOYS' で、前者なら σ/π 分離有り。 |
IFLOC |
'OCC' |
局在化リクエスト。'NO', 'NONE', 'OCC', 'VAC', 'BOTH' |
CHKFZC |
'NO' |
'YES' の場合、凍結内殻軌道を用いる。 |
LPRINT |
2 |
出力レベルの指定。2 ないし 3 を推奨。 |
Note
- CAFI 解析 を行うためには事前に SCC 計算後のモノマーの軌道係数を決定しておく必要がある。
- 解析には CPF ファイル (.cpf) も必要となるため同時に出力しておく。
- CPF のバージョンは
4.2を指定し、次のような入力で通常の FMO 計算を行い、CPF ファイルと SCC ファイル (.scc) を書き出しておく。
Example
1 2 3 4 5 6 7 8 9 | |
CCPT ネームリスト
多体相関計算を制御するためのネームリスト。 デフォルトでは MP4(SDQ)が実行される。
キーワード |
デフォルト |
説明 | |
|---|---|---|---|
METCOR |
'MPPT' | 相関法の指定。
|
|
MPLVL |
4 | Møller–Plesset 摂動の次数の指定。 | |
IFSNGL |
'YES' |
1 電子励起の寄与を含める指定。 | |
IFTRPL |
'NO' |
Full MP4 や CCSD(T) を行う場合、'YES' とする。計算コストが上がることに注意されたい。 |
|
THRENE |
1.d-6 | 結合クラスター展開、 2 次配置間相互作用などの繰り返し計算におけるエネルギーの閾値。 | |
THRRSD |
1.d-6 | 結合クラスター展開、 2 次配置間相互作用などの繰り返し計算における振幅ベクトルの残渣の閾値。 | |
MAXITR |
50 | 結合クラスター展開、 2 次配置間相互作用などの繰り返し計算における繰り返し回数の上限。 | |
IFBD |
'NO' |
結合クラスター展開でのブルックナー型 (BD) の指定。 | |
| MXBDIT | 10 | ブルックナー最適化回数の上限。 |
XYZ ネームリスト
ReadGeom に PDB ファイル等の構造ファイルを指定するのではなく、構造の座標を ajf ファイル内で直接指定する場合に用いられる。詳細は「座標の入力方法」を参照のこと。
FRAGMENT ネームリスト
FRAGMENT ネームリスト は、AutoFrag='OFF' もしくは AutoFrag='HYBRID' として、 全体もしくは部分的なフラグメント分割をユーザー指定で行う場合に用いられる。
以下の 5 つの項目を記述する必要があり、これらは BioStation Viewer を用いることで簡単に出力できる。
- フラグメント を構成する 原子の個数
- 8 桁右詰 (フォーマット は 10I8)。
- 10 フラグメントを超える場合は、10 フラグメント毎に改行して入力する。
- フラグメント の形式電荷
- 8 桁右詰。
- 10 フラグメントを超える場合は、10 フラグメント 毎に改行して入力する。
- 結合電子を自分のフラグメントに割り当てたフラグメント 間の結合の本数
- 8 桁右詰。
- 10 フラグメントを超える場合は、10 フラグメント毎に改行して入力する。
- フラグメント を構成する 原子の番号
- 8 桁右詰。
- 10 原子を超える場合は、10 原子毎に改行して入力する。
- 結合している相手のフラグメントの原子の番号 (8 桁右詰) 相手のフラグメントと結合している原子の番号 (8 桁右詰)
- 結合電子を自分のフラグメントに割り当てた、フラグメント間の結合の本数回繰り返し
- 用いる射影演算子の種類
2: sp2 射影演算子の適用箇所3: sp3 射影演算子の適用箇所- 無指定の場合には、3 が設定される (8 桁右詰)。
Example
具体例として、数種類のアミノ酸残基からなるペプチド(Ala-Asp-Gly-Lys-Gly-Glu)を 6 分割した場合 (次図) に対応する FRAGMENT ネームリストを示す。ピンク、青、緑、オレンジ、水色、紫の順でフラグメントの情報を記した。
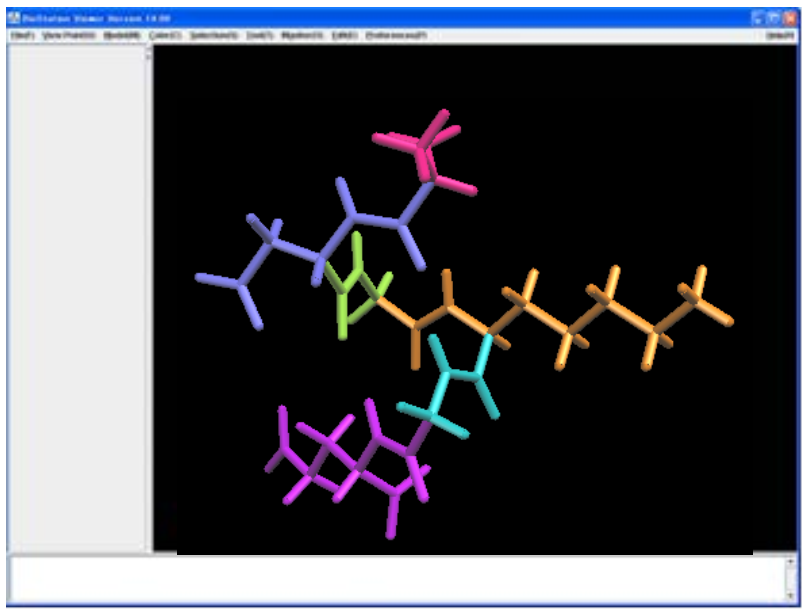
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 | |
Note
- 15 行目以降の末尾の
3は sp3 射影演算子を指定している。
MDCNTRL ネームリスト
MDCNTRL ネームリストは、 FMO-MD 計算の制御を行う入力。なお、ここでの FMO-MD は、 (F)MOMD だけでなく、 (F)MO-EM 機能も含まれる。 OPTCNTRL ネームリストで指定される、構造最適化とは別系統の実装なので、注意して欲しい。
FMO-MD を activate した場合、出力が大幅に増える(一点計算 × ステップ数)ので併せて注意。
キーワード |
デフォルト |
説明 |
|---|---|---|
MD |
'OFF' |
'ON' の場合、 MD/EM 計算を行う。 'OFF'の場合は一点計算になる。 |
(以下、 MD が 'ON' の場合のみ有効) |
||
IUP_RESTART |
5 |
restart ファイルの更新頻度。 |
TEMPLATE_AJF |
'template.ajf' |
restart.ajf ファイルを作るときの鋳型ファイルの名前。MDCNTRL ネームリストは、 1 行 1 パラメータになっていることが必要。 |
RESTART_AJF |
'restart.ajf' |
MD (EM) 計算の継続実行用入力ファイル。 TEMPLATE_AJF の入力パラメータを引き継ぎつつ、時刻、座標、速度、能勢粒子の座標と速度は、 MD ランの最終値が入る。 |
RDVEL |
'OFF' |
'ON' の場合、入力 ajf ファイルから、 速度 (VEL)を読み込む。 |
MIN |
'' |
極小化計算を指定する。
|
TIME_INIT |
0.0 |
MD の開始時刻 (ps) を設定。 |
TIME_STEP |
0.001 |
MD の時間ステップ (ps) を設定。 |
NSTEP |
10 |
MD/EM の全ステップ数。 |
ENSEMBLE |
'NVE' |
MD のアンサンブル。
|
RDNHC |
'OFF' |
'ON' の場合、NHC 粒子のデータ を ajf から 読み込む。 |
TEMP_INIT |
-9.999 |
初期温度 (K)。 負の値の場合は、 0 K (粒子に速度を与えずにMD を開始する)。 |
TEMP_SET |
298.0 |
定温 MD の設定温度 (K)。ENSEMBLE='NHC', ENSEMBLE='SCALING'の場合のみ有効。 |
CENT_FIX |
'OFF' |
'ON' の場合、重心座標を原点に固定する。 |
ROT_FIX |
'OFF' |
'ON' の場合、重心周りの回転運動を 0 にする。 |
SPHERE_RESTRAIN |
'OFF' |
'ON' の場合、分子系全体に球形の束縛ポテンシャルを掛ける。 |
SPHERE_CENTER |
'0' | SPHERE_RESTRAIN='ON' でのみ有効。束縛中心の原子のリスト。 例: '1-8,10' → 原子 1-8 および 10 の重心が設定される。 '0' の場合、 原点(0,0,0)に設定される。 |
SPHERE_RADIUS |
999.0 |
SPHERE_RESTRAIN='ON' でのみ有効。束縛半径 (Å) |
BLUE |
ブルームーン法。
|
|
RC_BLUE |
0.0 |
拘束距離(Å)。BLUE='RIJ', 'RIJ-RJK' のみで有効。 |
ICENT_BLUE |
'' |
重心 RI を構成する 原子。BLUE='RIJ', 'RIJ-RJK' のみで有効。例: '1-3' → 原子 1-3 の重心が RI になる。JCENT_BLUE, KCENT_BLUE も同様。 |
JCENT_BLUE |
'' |
重心 RJ を構成する 原子。BLUE='RIJ', 'RIJ-RJK' のみで有効。 |
KCENT_BLUE |
'' |
重心 RK を構成する 原子。BLUE='RIJ-RJK' のみで有効。 |
MODE_DYNFRAG |
0 |
Dynamic fragmentation (DF 動的フラグメント化)アルゴリズムのモード。
|
MODE_STATFRAG |
0 |
MODE_DYNFRAG > 0 でのみ有効。静的フラグメント (SF)のモード。
|
STATFRAG |
'0' |
MODE_STATFRAG > 0 でのみ有効。固定フラグメント SF を指定する。 例: STATFRAG='3,6,9' → 初期入力されたフラグメントのうち、 3、 6, 9 番は SF として扱われ、残りは DF として扱われる。 |
VEL ネームリスト
原子の速度を入力する ネームリスト。 MDCNTRL ネームリスト で、 MD='ON' かつ RDVEL='ON' の場合のみ、読み込まれる。
-
形式を以下に示す。
1原子の番号 元素記号 vx vy vz iopt
Example
-
例:
1 2 3 4 5 6 7 8 9 10 11 12 13
&VEL 1 F -0.0764963 -0.1224159 0.0885156 1 2 H 0.2271893 1.7095560 0.5821974 1 3 F 0.0288177 0.1128340 0.2193645 1 4 H -2.4459122 1.0800167 -0.9585628 1 5 F 0.0383582 0.2708645 -0.3835307 1 6 H 1.3436518 1.5684160 -0.7362045 1 7 H -1.5440933 -0.2595987 -0.2836240 1 8 F -0.2271486 0.0054176 0.1404151 1 9 H -0.0830465 -1.8152738 0.2779888 1 10 F -0.3755703 0.0308870 0.0944193 1 11 H -0.3986413 1.9783257 -0.4223578 1 /
Note
原子の番号と元素記号は、実際には読まれない。また、 iopt=0 は動かない原子、iopt=1 は動く原子だが、これも実際には読まれない。読まれるのは、実際の速度 vx, vy, vz で、これらは、AMBER 力場の内部単 位で出力される。
NHC ネームリスト
Nose-Hoover chains 法の熱浴粒子を入力する ネームリスト。 MDCNTRL ネームリスト で、 MD='ON' かつ RDNHC='ON' の場合のみ、読み込まれる。
-
形式を以下に示す。
1 2
mnhc 熱浴粒子の数(MDCNTRL ネームリストで指定された値よりも優先) j r_nhc(j) v_nhc(j) 粒子の番号、座標、速度
Example
-
例:
1 2 3 4 5 6 7 8
&NHC 5 1 -0.71143187E-02 -0.17370451E+00 2 -0.12977852E+00 -0.26274035E+01 3 -0.12467031E+00 -0.24234655E+01 4 -0.12485202E+00 -0.24347114E+01 5 -0.11981471E+00 -0.22408306E+01 /
TYPFRAG ネームリスト
DF で生成する 可能性のある フラグメント 種のデータ。MDCNTRL ネームリスト で、 MD='ON' かつ MODE_DYNFRAG > 0 の場合のみ、読み込まれる。
-
形式は以下に示す。
1フラグメント名(ただの記号) 形式電荷 構成原子群
Example
-
例:
1 2 3 4 5 6 7 8 9 10 11
&TYPFRAG OH -1 O H H2O 0 O H H H3O 1 O H H H O2H -3 O O H O2H2 -2 O O H H O2H3 -1 O O H H H O2H4 0 O O H H H H O2H5 1 O O H H H H H O2H6 2 O O H H H H H H /
座標の入力方法
- ABINIT-MP では原子の座標を入力する方法として、 以下の 2 通りの方法がある。
- CNTRL ネームリストの
ReadGeomで座標データファイルを指定する方法ReadGeomでは、 PDB フォーマットと MDL MOL フォーマットのファイルを指定することができる。
-
XYZ ネームリストで直接座標を指定する方法
-
ABINIT-MP では原子の座標を入力する場合に,以下の 2 つの入力方法が使用できる.
-
xyz 座標:
1原子の番号 元素記号 1 x 座標 y 座標 z 座標 最適化変数- xyz 座標は Å 単位で入力する.
- 原子間距離 の場合は入力エラーになる.
- 座標最適化変数が, 0 の場合はその変数は固定し, 1 の場合は最適化を行う.
- それぞれのデータは,データの間を 1 個以上の半角スペースで区切る.
-
内部座標
1原子の番号 元素記号 2 参照原子 1 参照原子 2 参照原子 3 最適化変数- は参照原子 3 からの距離(Å).
-
は結合角(参照原子 2-参照原子 3-入力原子)(度).
- (度) の範囲内で指定する.
-
は二面角(参照原子 1-参照原子 2-参照原子 3-入力原子)。
- の範囲内で指定する.
- 参照原子には,ダミー原子が使用できる.
- 最適化変数が, 0 の場合はその原子は固定し, 1 の場合は最適化を行う.
- それぞれのデータは,データの 間を 1 個以上の半角スペースで区切る.
-
- CNTRL ネームリストの
sp2 射影 BDA の使用方法
ABINIT-MP BINDS2019(Open Rev. 25) 以降では、 sp2 射影 BDA を併用することが可能となっている。使用方法は以下のとおりである。
自動分割の場合
-
sp3 分割、かつ sp3 射影 (通常計算)
1 2 3 4 5 6 7
&FMOCNTRL FMO='ON' Nbody=2 AutoFrag='ON' FragSizeAminoacid='+amino' MainProj='SP3' : (省略) -
sp2 分割、かつ sp2 射影
1 2 3 4 5 6 7
&FMOCNTRL FMO='ON' Nbody=2 AutoFrag='ON' FragSizeAminoacid='+peptide' MainProj='SP2' : (省略)Note
MainProjですべての射影演算子の種類を一括設定する。- デフォルトは、
MainProj='SP3'であり、全てに sp3 射影演算子が設定される (sp2 分割の際のデフォルトはMainProj='SP2'となる)。 MainProj='SP2'とすると、全てに sp2 射影演算子が設定される (秋永版と同様の動作)。- 6-31G* 以外ではエラー終了するようになっている。
LMOTYPの指定によらず、sp2 ではPOPUで作成したものが適用される (後述する手動分割 or ハイブリッド分割で射影演算子を併用する際には sp3 は ANO としてLMOTYPを併用可能にするため)。FragSizeAminoacidとMainProjは独立に指定可能 (4 通り)
手動分割の場合
-
全て sp2 射影
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
&FMOCNTRL FMO='ON' Nbody=2 AutoFrag='OFF' MainProj='SP3' LMOTYP='ANO' NF=5 : (省略) &FRAGMENT 8 7 7 7 9 1 0 0 0 -1 0 1 1 1 1 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 3 9 2 11 16 2 18 23 2 25 30 2 /Note
- 適用順序:
MainProjで指定された射影演算子が全体に指定される。- 18 行目以降 (フォーマットは、
BDA BAA Proj) が Proj の値で上書きされる。&FRAGMENTで Proj の指定が無い場合は、MainProj の値が保持される。
- LMOTYP` は sp3 射影演算子のみに適用される (sp2 射影演算子は常に POPU)。
- 一部でも sp2 射影が指定されている場合、6-31G* 以外ではエラー終了する。
- 適用順序:
-
全て sp3 射影
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
&FMOCNTRL FMO='ON' Nbody=2 AutoFrag='OFF' MainProj='SP3' LMOTYP='ANO' NF=5 : (省略) &FRAGMENT 8 7 7 ... 1 0 0 ... 0 1 1 ... 1 2 3 ... 9 10 11 ... 16 17 18 ... 23 24 25 ... 30 31 32 ... 3 9 11 16 18 23 25 30 /Projが無ければ、MainProjが適用される。
-
sp2/sp3 射影併用 (
MainProj='SP3'の場合)1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
&FMOCNTRL FMO='ON' Nbody=2 AutoFrag='OFF' MainProj='SP3' LMOTYP='ANO' NF=5 : (省略) &FRAGMENT 8 7 7 ... 1 0 0 ... 0 1 1 ... 1 2 3 ... 9 10 11 ... 16 17 18 ... 23 24 25 ... 30 31 32 ... 3 9 11 16 2 18 23 2 25 30 /- 同じ設定として計算が実施される。
MainProjと同じ 2 や 3 は省略可能。
-
sp2/sp3 射影併用 (
MainProj='SP2'の場合)1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
&FMOCNTRL FMO='ON' Nbody=2 AutoFrag='OFF' MainProj='SP2' LMOTYP='ANO' NF=5 : (省略) &FRAGMENT 8 7 7 ... 1 0 0 ... 0 1 1 ... 1 2 3 ... 9 10 11 ... 16 17 18 ... 23 24 25 ... 30 31 32 ... 3 9 3 11 16 18 23 25 30 3 /- 同じ設定として計算が実施される。
MainProjと同じ 2 や 3 は省略可能。
指定された射影演算子の確認方法
-
以下の設定を例とする (sp2/sp3 射影併用)。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
&FMOCNTRL FMO='ON' Nbody=2 AutoFrag='OFF' MainProj='SP3' LMOTYP='ANO' NF=5 : (省略) &FRAGMENT 8 7 7 7 9 1 0 0 0 -1 0 1 1 1 1 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 3 9 11 16 2 18 23 2 25 30 / -
ログファイルに以下のように書き出される。
1 2 3 4 5 6 7 8 9 10 11 12 13
## MONOMER ONE-ELECTRON INTEGRAL ## SET MONOMER PROJECTION OPERATOR ## SET PROJECTION OPERATOR: SP3 3 ## SET PROJECTION OPERATOR: SP2 11 ## SET PROJECTION OPERATOR: SP2 18 ## SET PROJECTION OPERATOR: SP3 25 ## MONOMER INITIAL MO&FMOCNTRL -
`LMOTYP='ANO' なので、sp3 射影演算子は ANO、sp2 射影演算子は POPU の併用となる。
- 全て POPU に揃える場合には、
LMOTYP='POPU'とする。
ハイブリッド分割の場合
-
例: HIV_P のリガンド LPV をハイブリッド分割
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27
&FMOCNTRL FMO='ON' AutoFrag='HYBRID' HybridFrag='200' HybridNF=4 MainProj='SP3' LMOTYP='ANO' : &FRAGMENT 26 20 27 21 ... 1 0 0 -1 ... 1 0 0 1 ... 3132 3133 3134 3135 ... 3153 3154 3155 3161 ... 3168 3169 3170 3223 ... 3156 3157 3158 3259 ... 3176 3177 3178 3179 ... 3142 3143 3144 3145 ... 3152 3186 3187 3188 ... 3195 3201 3202 3203 ... 3196 3197 3198 3199 ... 3212 3213 3214 3215 ... 3222 3154 3156 2 3159 3186 3195 3196 /Note
HybridFragでは、手動分割したいフラグメントの "自動分割時に付与されるフラグメント番号" を指定する (LPV は 200 番)。HybridNFでは、HybridFragで指定したフラグメントを何分割するか指定する。HybridNFの数だけ、&FRAGMENTの情報が読み込まれるので、&FRAGMENT には、LPV の分割情報のみを記述する。&FRAGMENTの BDA-BAA-Proj のペアは、MainProj=3 の場合には sp2 にしたい部分だけ記述すればよい。LMOTYP='ANO'なので、sp3 射影演算子は ANO、sp2 射影演算子は POPU の併用となる。全て POPU に揃える場合には、LMOTYP='POPU'とする。