











研究内容
日本発の理論手法であるフラグメント分子軌道(FMO)法は、電子状態計算に基づいて化合物と標的タンパク質との詳細な相互作用情報が得られることから、従来法と比較して飛躍的に精度の高い信頼性と有効性をもった論理的創薬に繋がることが期待されています。FMO法は、1999年に北浦教授らによって提案されて以来、基礎および応用の両面で多くの研究がなされてきました。これを実用的な創薬技術として発展させるためには、アカデミアと産業界との密接な連携に基づいた研究開発が不可欠です。2014年11月に発足した「FMO創薬コンソーシアム」は、High Performance Computing Infrastructure (HPCI)などを活用して、FMO法のインシリコ創薬への活用を一層加速するため、FMO研究者が企業の創薬研究者とともに実践的応用計算手法の開発と普及を進める組織であり、皆様に広くご参加を呼びかけています。本コンソーシアムでは、FMO法を有効に活用して、
- タンパク質とリガンド分子の結合特性予測、リガンドスクリーニング
- ドッキング、分子動力学、バイオインフォマティクス等のインシリコ手法との連携
- 構造生物学と連携した構造データの解釈およびFMO法を援用した構造精密化
- FMOデータベースの構築
- 大規模データに基づく機械学習やAI予測
- FMO力場の開発
などを皆様方と協力して行い、計算・解析手法のノウハウと計算結果データの共有を行います。そして、参画企業研究者の皆様には、現場の創薬計算を想定したアドバイスや計算手法・結果に対する客観的な評価を行っていただき、広くご意見を伺える場にしていきたいと考えております。
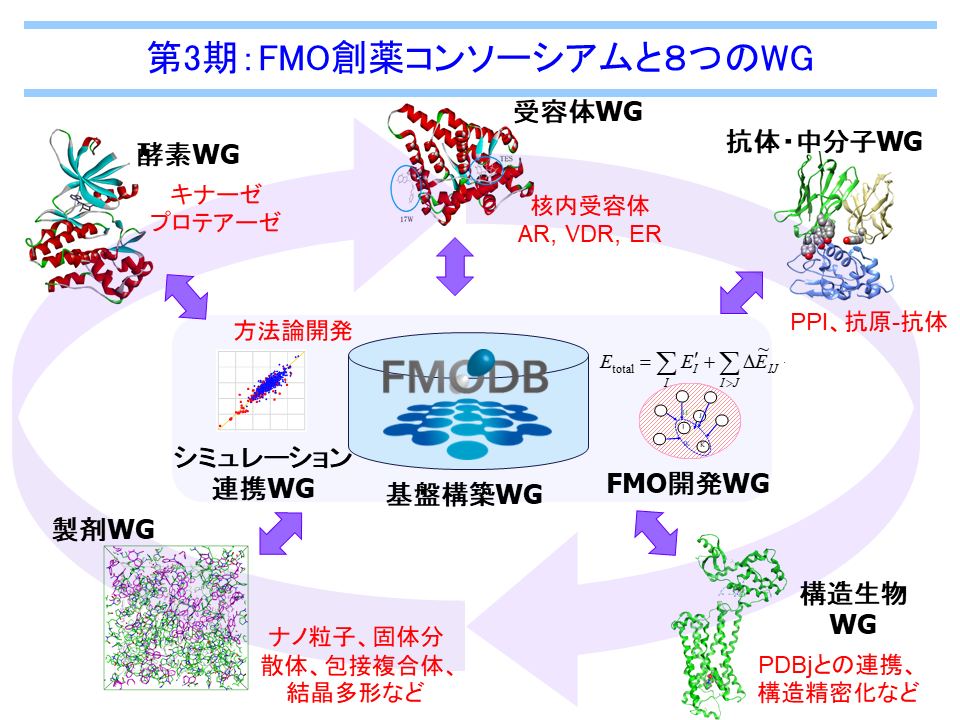
WG紹介
FMODDでは、3つの創薬ターゲットWG、2つの創薬関連WG、3つの創薬手法開発WGに分かれて活動を行い、それぞれのWGで検討した成果を持ち寄って全体の議論を行っています。各WGではアカデミアの取りまとめ役を中心として産学官のメンバーが参加しています。
酵素 WG
-
 田中 成典神戸大学大学院
田中 成典神戸大学大学院 -
 矢城 陽一朗岡山理科大学
矢城 陽一朗岡山理科大学
キナーゼ
p38 MAPキナーゼやJAKキナーゼ等をターゲットとした活性化合物の結合エネルギーの検討や、ターゲット特異性を評価しています。p38 MAPキナーゼは、FMODDの中で最も多様なリガンドについて検討した事例となっています。95種類のp38-リガンド複合体構造について、IC50とFMO相互作用エネルギーとの相関を検討したところ、全体では相関がみられませんでしたが、化合物を骨格ごとに分類することや特異値分解の手法を利用することで、相関関係を乱す「ノイズ」を取り除き、相関係数の値が改善することがわかりました。モデリング手順から計算結果の分類方法などのノウハウをまとめた論文[Sheng et al., 2018]や、IFIE行列の特異値分解によって実験値との相関を改善した論文 [Maruyama et al., 2018] を発表しています。
ヤーヌス・キナーゼ(JAK) はJAK1, JAK2, JAK3, Tyk2の4つのサブタイプをもち、FMOによるターゲット選択性の評価を行っています。また、キナーゼの特徴である、リン酸化チロシンのリン酸化状態によるリガンド結合性への影響を評価しています。さらに、機械学習によるIFIE値の予測等の検討を進めており、この研究に関しては論文発表[Tokutomi et al., 2020]を行いました。
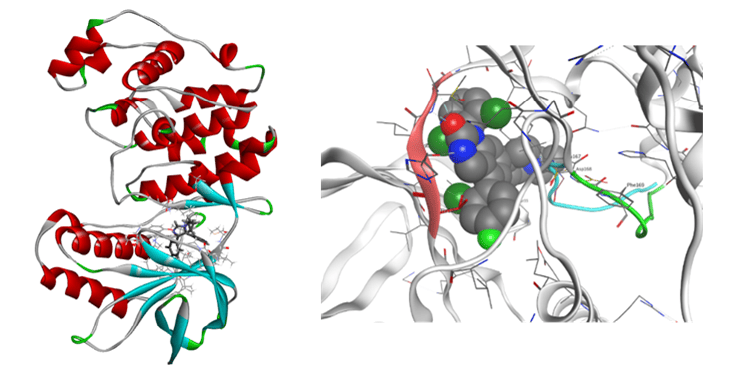
プロテアーゼ
レニン-阻害剤系やHIV-1プロテアーゼ、HIV逆転写酵素などに対するFMO計算解析を行い、IC50や他の実験値、副作用との比較、検討を行っています。20種のレニン-阻害剤複合体と、9種のHIV-1プロテアーゼ-阻害剤複合体に対するFMO計算の結果では、Total IFIE(結合エネルギー)と活性値(IC50値およびKD値)との間に強い相関が認められました。また、HIV-1プロテアーゼの活性中心アミノ酸残基のプロトン化状態を考慮した計算の結果、中性子回折で観測されたプロトン化状態を再現しました。加えて、HIV-1プロテアーゼの各アミノ酸残基と阻害剤のIFIE解析から、活性相関に関与するアミノ酸残基を特定できる可能性を強く示唆する結果を得ました。
さらに、HIV-1非核酸系逆転写酵素-阻害剤複合体に対して構造活性相関の解明およびHIV-1変異株に有効な新規誘導体の開発に取り組んでいます。
受容体WG
-
 栗田 典之豊橋技術科学大学
栗田 典之豊橋技術科学大学
アンドロゲン受容体(AR)、ビタミンD受容体(VDR)、レチノイン酸受容体(RORgt)、及びエストロゲン受容体(ER)などと様々なリガンド間の相互作用をMD及びFMO計算を用いて解析しています。各リガンドの活性値の評価、受容体に強く結合する新規化合物の提案、activity cliffの原因解明、作動薬あるいは逆作動薬の作用機序相違の原因解明、サブタイプ選択性の評価、などを検討しています。
ARでは、既存のリガンドとAR間の相互作用を解析し、その結果を基にARにより強く結合する化合物を新規のARリガンドとして提案しました[Kobayashi et al., 2017]さらに、上位の既存リガンドの官能基に4種類の置換基を導入した新規リガンドを提案し、ARとの結合特性を解析することで、よりARとの結合親和性の高い新規リガンドを得ることができました[Nakamura et al., 2021]。
VDRでは、化学構造が同じでキラリティのみが異なる2つのリガンドの活性値が大きく異なる原因を、リガンド周辺に存在する2つのHis残基のプロトン化状態の変化を考慮することにより、初めて明らかにしました[Terauchi et al., 2019] 。RORgtでは、作動薬、あるいは逆作動薬がRORgtに結合した際の構造変化をMD計算により解析し、転写活性に重要であるHelix12の構造変化の相違を明らかにしました。その結果は、既知の作動薬と逆作動薬の作用機序の相違を矛盾なく説明できました。さらに、MD計算で求めた幾つかの特徴的な構造に対し、FMO計算によりRORgtのアミノ酸残基とリガンド間の相互作用を解析し、構造変化のトリガーとなるHis479周辺の相互作用変化を明らかにしました[Suzuki et al., 2020] 。
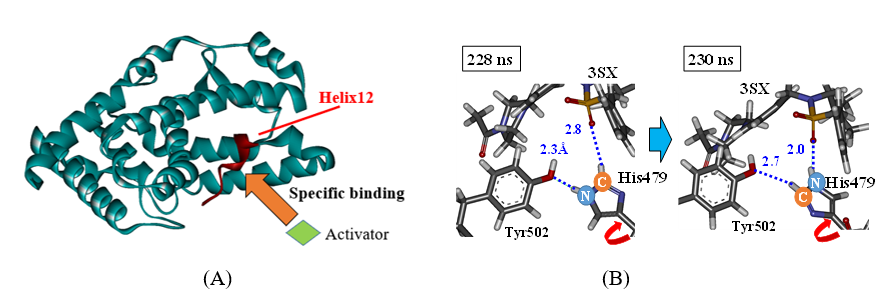
Figure (B) His479側鎖の回転によるRORgtと逆作動薬3SX間の相互作用変化
ERでは、2つのサブタイプ(ERα、ERβ)に対してPDBに登録されているリガンドとの複合体構造を網羅的に計算しました。IFIEおよびPIEDA相互作用に基づくクラスタリング解析(VISCANA)から、ER-リガンド間の相互作用の特徴とサブタイプ選択性との関係を検討し、リガンド結合に必須なアミノ酸残基とβ選択性に関与する残基が明らかとなりました[Seki et al., 2018]。
抗体・中分子 WG
-
 川下 理日人近畿大学
川下 理日人近畿大学
理工学部
生命科学科
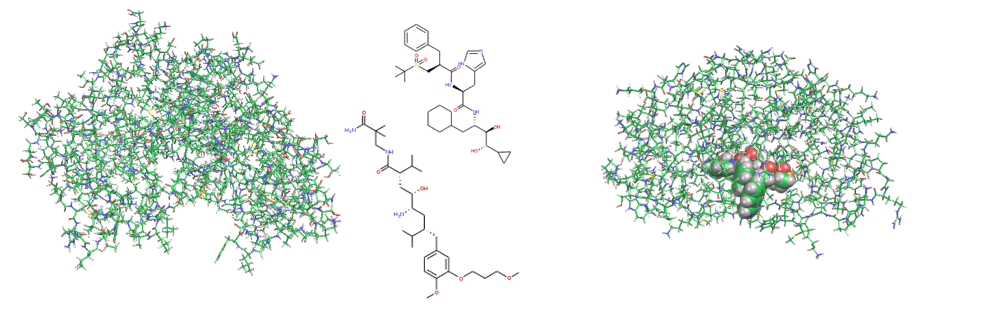
ブロモドメイン、βセクレターゼ、TCR-pMHC、FimH-マンノース誘導体、MDM2-p53、HIV gp120-anti gp120抗体系などの多様なタンパク質-タンパク質間相互作用を扱い、薬剤耐性の評価や結合予測、抗体医薬開発に繋がる抗原抗体反応についても検討しています。AIによる活性予測も行っています。
ブロモドメイン系では、ペプチドリガンドとフラグメント化合物との相互作用を比較検討し、阻害剤のデザイン過程における相互作用の変化について理論的に解析しました。5つのリガンド化合物とIC50との相関はR2=0.82という良好な値であり、結合親和性が増加するにつれて、フラグメント化合物がペプチドリガンドのIFIEと類似の相互作用を獲得することが明らかとなりました。さらにCH/π相互作用、電荷の変化やリガントと水分子との相互作用について解析しました[Ozawa et al., 2017a, b]。
TCR-pMHC系では、免疫学研究において重要な、T細胞受容体(TCR)とペプチド–主要組織適合複合体(pMHC)との特異的/非特異的認識についてCH/π相互作用に着目した検討を行いました。各共結晶被提示ペプチド、2, 3, 5, 8の位置に7, 8, 2, 3種の変異を入れた構造及び野生型105(21 X 5)構造に対してPIEDA計算を行い、各CDRとペプチド間の相互作用を明らかにしました[Tsuchiya et al., 2018]。
SARS-Cov-2 Spike蛋白のRBD(抗体結合部位)とペプチド抗体医薬との相互作用計算を行い、T415, K417, Y421, F456, A475, F486, N487, N501, and Y505が、相互作用に重要な働きを持つことが示されました。このうち、K417, N501は、感染性、病原性に影響があるとされている変異株が報告されており、相互作用の変化(特に減少)が、感染性、病原性の重要な因子になっていることが推察されます[Watanabe et al., 2021]。
PPIに関するこれらのFMO計算から、主として水素結合が特異性の高い認識に、CH/π結合が非特異的認識に深く関与している可能性が示唆されています。
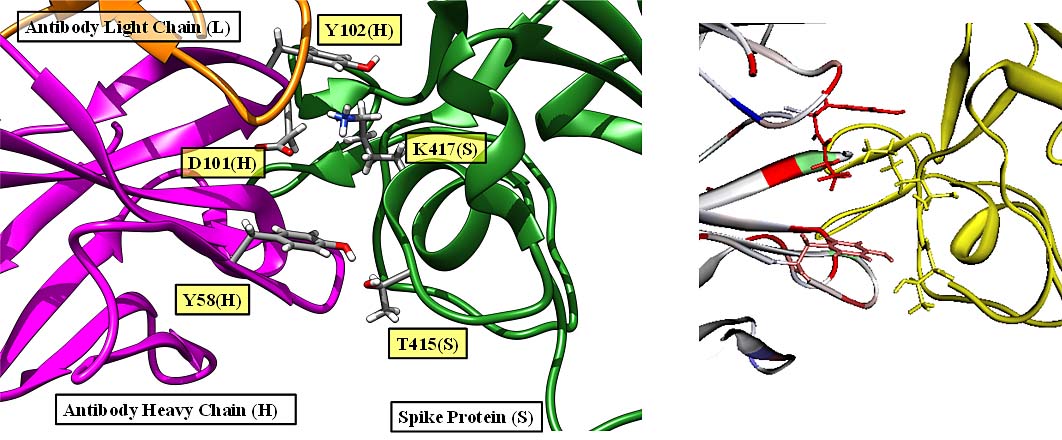
(左)PDBID:7CH5、(右)FMODBID: JM5M9
構造生物WG
-
 上村 みどりCBI研究機構量子構造生命科学研究所
上村 みどりCBI研究機構量子構造生命科学研究所
第3期より加わった構造生物WGは以下のテーマに特化して構造生物学領域と計算科学領域の融合を目的として、以下の優先順位に従い研究を進めます。FMO計算にしても製薬会社にもメリットがあるターゲットを計算手法自体の確立していくようなプロセスが含まれるターゲットを選択しています。
⑴ FBDD using FMO (X線解析、NMRの結果の計算科学による説明)
構造生物領域からもリクエストが多いメタロプロテアーゼについて、LpxCを例に計算を実施していきます。メタロプロテアーゼについてのFMO適応は、FMODDの共通の課題でもあります。まだ確立した手法がない領域であり、配位子のプロトネーションについても様々な場合を考慮すべきであり、難しい課題ではありますが、FMO開発WGとも密に連携し、チャレンジしたいと思います。阻害活性値(IC50)とFMO計算によるIFIEやtotal energyとの相関を説明できるようにしたいと思います。もう一つの具体例としては、PPIを安定化する化合物のFBDDを進めます。アダプタータンパク質14-3-3を介した複数のタンパク質との相互作用を安定化する化合物(Stabilizer)となるという活性を有した化合物デザインです。 PROTACSの点からも注目のターゲットと考えています。
⑵ Antibody/Peptide からsmall molecule へのreduction (X-ray+Cryo-EM)
ペプチドや抗体の低分子化は、至上の命題でもありますが、なかなかうまくいっていません。テーマ設定においては、WG内で募集した結果、IL2低分子化を採用することにしました。他にも、良質なターゲットを探索して進めていきます。
⑶ 膜タンパク質 (X-ray, Cryo-EM)
まだ具体的なターゲットは選定中ですが、GPCRを中心に実施していく予定です。
⑷ 構造生物学領域研究者との連携
日本蛋白質構造データバンク(PDBj)および昨年度設立されたCBI研究機構量子構造生物学研究所との連携も含め、構造生物学領域の研究者とのミーティングやセミナーを通じて連携を強化して創薬研究課程に直接役立つような取り組みを進めていきます。
製剤WG

-
奥脇 弘次JSOL, 大阪大学
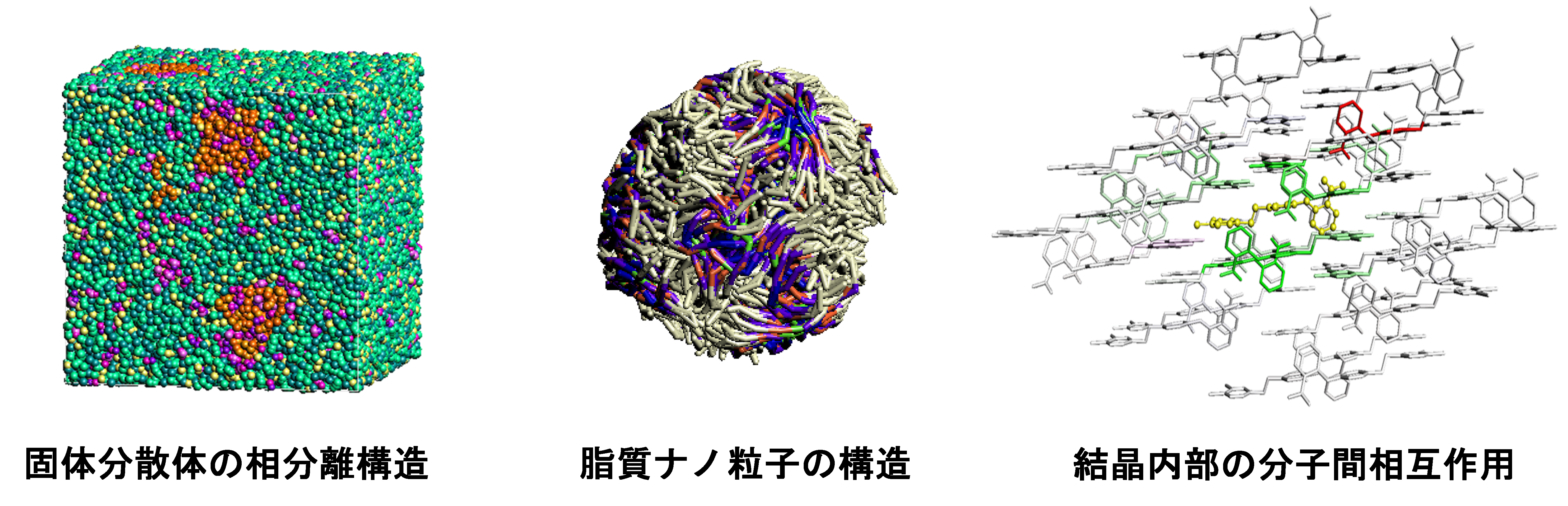
製剤WGでは、原薬の製剤化に関する分子論的な検討をFMO計算に基づいて行い、量子化学計算による分子間相互作用を製剤設計に活用する道を切り拓くことを目的としています。FMO-DPD計算による相分離挙動やナノ粒子形成過程の解明、全原子MD計算による構造および物性の評価、FMO計算による分子間相互作用の定量的評価など、最適な製剤設計に向けたシミュレーション技術を開発しています。製剤WGの活動は実験的研究と共同で行い、相互検証を繰り返すことで実用的な技術開発に繋げます。現在、4つのテーマを推進中です。
⑴ 結晶安定性の評価とコフォーマーのスクリーニング
⑵ 固体分散体の分子分散状態の評価と相分離挙動の解明
⑶ 脂質ナノ粒子の設計
⑷ 包接複合体の分子挙動の解明
シミュレーション連携WG
-
 加藤 幸一郎九州大学
加藤 幸一郎九州大学
古典MDやドッキングなどのシミュレーションとFMOの連携を目指したWGです。これまでにKBDDコンソーシアムと連携し、タンパク質-リガンド水和複合体の多数のMDサンプリング構造[Araki et al., 2016]に対してFMO計算を行い、それらに基づく高精度な活性予測法の開発などを行ってきました([Mochizuki et al., 2021]のFukuzawa et al. “FMO Drug Design Consortium” を参照)。現在はAIの活用にも取り組んでおり、ライフインテリジェンスコンソーシアム(LINC)のPJ14と連携して、FMO法と機械学習を組み合わせた高精度分子力場の開発に向けた検討や相互作用予測モデルの開発などを進めています。FMO法を用いることで、タンパク質-リガンド複合体の様な巨大ヘテロ系の高精度学習データを作成することが可能となるだけではなく、IFIEやPIEDAを得ることができます。これらの優位性を活かしたシミュレーションとの連携を世界に先駆けて進めており、既存分子力場の高度化を目的としてFMO法で得られたRESP原子電荷を高精度に予測可能な機械学習モデルの開発にも成功しました [Kato et al., 2020]。
FMO開発WG
-
 中野 達也国立医薬品食品衛生研究所
中野 達也国立医薬品食品衛生研究所
FMO専用プログラムABINIT-MP およびBioStation Viewer などの基盤ソフトウェアの整備・開発を行っています。FMO法による溶媒効果の考慮や効率的な構造最適化など、創薬のためのFMO方法論を開発します。
タンパク質–リガンド結合能の評価において、リガンド周辺の量子力学的な構造緩和が活性相関を高めるため、FMO創薬の実用化に向けた重要な課題として最適化技術の開発を行ってきました。リガンドとその周辺のみを緩和領域とするFMOベースの部分構造最適化をコア技術として、これまでAMED-BINDSプロジェクトと連携してフローズンドメイン法ならびに冗長座標変換の導入による高速化を試みています。こうした部分構造最適化技術の実用化に向けて、種々のタンパク質–リガンド複合体系をターゲットとした検証に取り組んでいます。また、生体内環境を記述する連続誘電体モデルを結合させたFMO計算の高速化・高度化、あるいは環境場を含めた構造最適化技術の開発も進めています。利便性の向上を図るため、自動フラグメント化機能における核酸を含めた対応残基種の拡張等も進めて行きます。
FMO計算で必要となるフラグメント分割やフラグメント間相互作用解析など、ABINIT-MP特有の入出力様式に対応した入力データ作成や結果可視化を可能とするBioStation Viewerの開発を行っています。ABINIT-MPの開発に対応した機能拡張も進めています。BioStation Viewerは本ホームページで無償公開しています。 https://fmodd.jp/biostationviewer-dl/
Molecular operating environment (MOE)は独自言語Scientific vector language (svl)をベースに高度に拡張可能な高機能分子モデリングソフトウェアです。MOE上でFMO計算ワークフロー(入力ファイル作成・結果可視化)を完結可能なFMOeの開発を行っています。入力ファイル作成インターフェースではタンパク質の自動フラグメント化および手動でのフラグメント化に対応し、可視化インターフェースではFMO計算結果より得られるIFIEやPIEDAをインタラクティブに可視化する事が可能です。
基盤構築WG
-
 本間 光貴理化学研究所
本間 光貴理化学研究所
FMOデータベースの開発や活性データの収集、煩雑な計算を自動処理するFMO自動計算プロトコールの開発、MDサンプリングのデータフローなど、創薬の為の研究基盤の整備を行うとともに、AI技術と連携して、後付けではなく新規な医薬候補品を創成する手法の開発を目指しています。
現在開発を進めている具体的な開発項目は以下になります。()内は、主な開発機関。
・FMO自動計算プロトコール(理研)
・FMOデータベース(FMODB、理研、星薬大)
・FMO-AIによる活性予測(理研、住友化学)
・FMO gridとその応用(理研、甲南大学)
・FMO力場(理研、九大、LINC)
FMO自動計算プロトコール: リガンドを含むタンパク質構造の欠損補完、プロトン化・脱プロトン化、互変異性、構造最適化、FMO計算用インプットの作成、計算実施、計算結果からの相互作用データの整形までを半自動的に行うプロトコールです[Watanabe et al., 2019]。熟練したFMO研究者の手動による計算と遜色ない精度での計算を実施できることをERαやp38 MAPキナーゼを用いて確認しており、1万を超えるFMODBのデータの過半数は本プロトコールによる計算結果になっています。プロトコールは、市販ソフトウェアの機能を利用していまずが、現在、無償化を進めています。
FMOデータベース(FMODB): FMODBは、世界で初めてのタンパク質の量子化学計算結果を収載したデータベースで [Takaya et al., 2021]、2019年2月に一般公開し、2020年3月にはCOVID-19関連タンパク質のデータを公開してプレスリリースを行いました。2021年4月現在で13,050件のデータを収載しています。現在、構造生物WGやPDBjと連携して高分解能なタンパク質構造の計算を系統的に実施しています。FMODB Home Page – https://drugdesign.riken.jp/FMODB/。
FMO-AIによる活性予測、FMO gridとその応用: FMO-AIは、多様なリガンドとタンパク質の複合体構造群のFMO計算の結果得られる相互作用のエネルギー値を用いて機械学習を行い、高精度の活性予測モデルを構築する手法です。学習に用いる記述子としては、PIEDAの他、相互作用エネルギーの3D配置を反映した記述子も開発しています。その中の一つが、FMO gridであり、ポケット内の格子点にプローブを配置して予め相互作用エネルギーを計算します。
FMO力場: シミュレーション連携WG、LINCと連携して、従来のAMBERベースの分子力場よりも量子力学的な相互作用を精密に再現できる新しい力場の開発を行っています。すでに、FMO電荷を再現するAI予測モデルを開発し、論文公開しています[Kato et al. 2020]。そのAI予測モデルのために数万個に上るFMO計算を行い、それらのデータの一部はFMODBにも公開されています。
フラグメント分子軌道(FMO)法
タンパク質や核酸等の生体高分子の量子化学計算は、計算量が膨大であるため、そのまま全体を解くことができません。FMO法では、巨大分子を小さなフラグメントに分割し、フラグメントとフラグメントペアの電子状態を周辺からの環境静電ポテンシャル存在下で解き、それらを組み合わせることで全体の電子状態を構築します。同時に、フラグメント間の相互作用エネルギー (Inter-Fragment Interaction Energy; IFIE、またはPair Interaction Energy; PIE) を得ることができるため、適切なフラグメントを設定することによって、分子内・分子間の相互作用を定量的に評価することができます[1] [2] [3]。
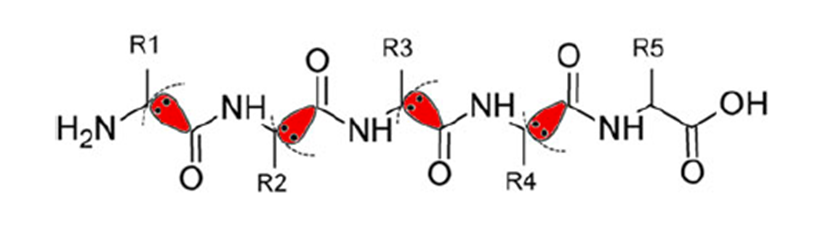
インシリコ創薬分野では、主にIFIEやそのエネルギー成分分割(Pair Interaction Energy Decomposition Analysis; PIEDA)を用いて、リガンドと標的タンパク質との結合(自由)エネルギーや分子間相互作用の評価を行っています。溶媒や構造の熱揺らぎの効果も適宜取り込んでいます。
[1] K. Kitaura et al. (1999) Chem. Phys. Lett. 313, 701-709 https://doi.org/10.1016/S0009-2614(99)00937-9
[2] D. G. Fedorov et al. (2012) Phys. Chem. Chem. Phys. 14, 7562-7577. https://doi.org/10.1039/C2CP23784A
[3] S. Tanaka et al. (2014) Phys. Chem. Chem. Phys. 16, 10310-10344. https://doi.org/10.1039/C4CP00316K
HPCIの利用
HPCI(High Performance Computing Infrastructure)は、スーパーコンピュータ「富岳」と全国の大学・研究機関の計算資源を高速ネットワークで結んだシステムです。FMODDでは、2015年度から産業利用課題「HPCI を活用したFMO 創薬プラットフォームの構築」を継続しており、これまでに「京」「富岳」(理化学研究所)、TSUBAME3.0(東京工業大学)、FX100(名古屋大学)を活用した研究を推進中です。
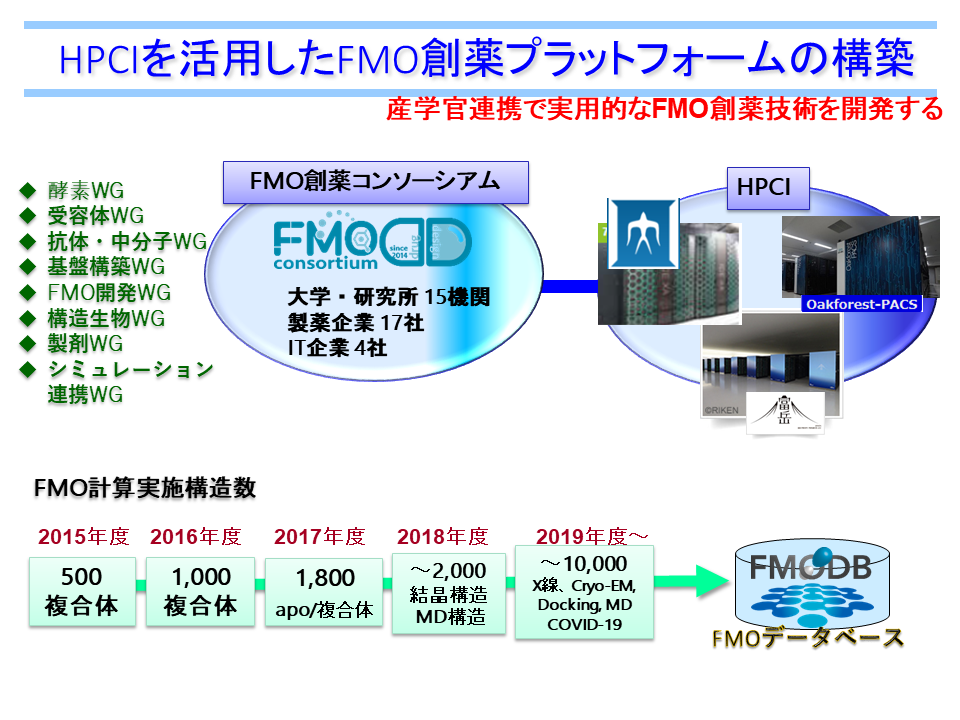
座談会「HPCIを語る」の動画が富岳百景のページから公開されました。
福澤代表がHPCIとFMO創薬コンソーシアムについて解説しています。